運動品牌Puma成功撤銷波蘭Sinda Poland鞋廠的歐洲共同體近似商標申請

歐盟普通法院(The General Court of the European Union)於今年(2016)2月25日針對運動品牌Puma SE(以下簡稱Puma)提出之商標異議做出了判決。本判決認為,Puma所擁有的美洲獅商標(詳參圖1、圖2),與其異議之波蘭鞋廠Sinda Poland Corporation sp.(以下簡稱Sinda Poland)(詳參圖3)申請中的商標確有相似。此判決翻轉了歐盟內部市場協調局(Office for Harmonization in the Internal Market,以下簡稱OHIM) 所作出兩商標不近似之決議。本判決中,歐盟普通法院判決認為,OHIM應撤銷Sinda Poland之商標申請,並命令Sinda Poland及OHIM支付Puma相關訴訟費用。
本爭議起始於2012年,波蘭鞋廠Sinda Poland申請一歐洲共同體商標(Community Trade Mark,簡稱CTM),該商標為一跳躍的貓科動物形狀(詳參圖3),並指定使用於鞋類及運動鞋類產品。OHIM於2012年9月公告Sinda Poland申請的商標,同年11月Puma向OHIM提起異議,表示Sinda Poland之商標(詳參圖3)與Puma之美洲獅商標(詳參圖1、圖2)過於近似,有令消費者混淆商品或服務來源之虞。然而,OHIM於2014年決議認為,Sinda Poland之商標係兩種跳躍動物之結合,形成獨特的動物圖案設計,與Puma之美洲獅商標不相似,應不具有混淆消費者的疑慮。
Puma向歐盟普通法院上訴,認為法院應撤銷Sinda Poland之商標申請,並要求Sinda Poland支付相關訴訟費用。歐盟普通法院於今年2月25日做出決議,認為Sinda Poland侵害了Puma之商標權,並指出OHIM未將系爭兩商標在視覺上及概念上之整體相似度列入考量。該法院認為「Puma與Sinda Poland之商標皆為黑色的動物剪影,兩商標之動物背部及腹部弧度雖有所不同,但兩商標無法否認地有相似之處。」
經過為期將近四年的爭訟,Puma成功撤銷了Sinda Poland近似商標之申請。對國際知名運動品牌如Puma而言,近似商標的存在將會造成品牌知名度及商譽的打擊。如何藉由排除近似商標之申請以鞏固品牌形象及消費者認同,對品牌企業而言是必須面對的重要課題。本案中,Sinda Poland之近似商標仍在申請階段時Puma即提起異議。Puma積極的維權行動,是品牌廠商能夠藉以參照的模範。

圖1 Puma跳躍美洲獅商標(註冊於尼斯分類第1-42類)(圖片來源:歐盟普通法院判決)
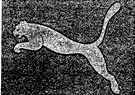
圖2 Puma跳躍美洲獅商標(註冊於尼斯分類第18、25、28類)(圖片來源:歐盟普通法院判決)

圖3 Sinda Poland欲申請之商標(註冊於尼斯分類第25類) (圖片來源:歐盟普通法院判決)
「本文同步刊登於TIPS網站(https://www.tips.org.tw)」

陶思妤
法律研究員 編譯整理
General Court ends Puma cat fight,Feb.25,2016 http://www.worldipreview.com/news/cjeu-ends-puma-cat-fight-9619?success_subscription=1(last visited :Mar.7,2016)
Puma SE v Office for Harmonisation in the Internal Market (Trade Marks and Designs) (OHIM),Mar.7,2016 http://curia.europa.eu/juris/document/document.jsf?text=&docid=174582&pageIndex=0&doclang=FR&mode=req&dir=&occ=first&part=1&cid=860012(last visited :Mar.7,2016)



一、立法背景 由於美國國家海洋暨大氣總署(National Oceanic and Atmospheric Administration,縮寫NOAA)於2018年間發布關於氣候變遷將導致經濟發展受到影響之相關報告,同時間,美國最高法院拒絕駁回2015年由21位民眾及美國Our Children’s Trust(非政府組織)對聯邦政府所提起之訴訟,主張美國政府並未循正當法律程序,即鼓勵對環境保護傷害甚鉅之石化能源開發。因此聯合國人權暨環境特別報告(UN Special Rapporteur on human rights and the environment)呼籲各國盡快針對環境變遷採取相關行動,美國國會議員Ed Markey及Alexandria Ocasio-Cortez遂基於上述情事於2019年2月7偕同提出綠色經濟草案(下稱本草案)。 二、草案簡介 所謂綠色經濟,是因應全球經濟危機、氣候變遷、石油資源枯竭而提出,其內容包括金融及租稅政策的重建以及再生能源的運用,初始概念於2007年由一位記者刊載於時代雜誌與紐約時報,後相關倡議人士遂依此成立非政府組織The Green New Deal Group,並於2008年廣泛發行相關刊物。 三、草案內容 本草案賦予政府五大義務:溫室氣體零排放、創造百萬高薪工作機會、投資基礎設施及工業、永續環境(諸如確保空氣、水質、氣候、食品之安全、韌性社區之推動)、反壓迫等,且內容上更將前開義務再行細分為14項目標計畫,並訂定10年執行期間。 上揭14項目標計畫的內容大致可分為五類,分別為:提升基礎設施以因應各種氣候變遷所造成之災害、將政府所需能源全數轉換為零碳排放、提升電力及能源效率、消除製造業與農業所造成之汙染與溫室氣體的排放,另外亦全面將大眾運輸設施改建為高速及零碳排放系統。 為達成前述14項目標,本草案一共訂定15項須政府配合之細項,方向上包括:給予社區、組織、機關、地方政府及各法人相關協助、提供適切之訓練課程及高等教育、針對新興科技之研究與開發進行投資、提高家庭所得及保障各級勞工組織工會之權利、提供全民高品質之健康照護。
2004年WIPO推出國際專利電子申請系統且申請數量激增世界智慧財產組織 (WIPO)於今年3月報導指出:WIPO於2004年推出了新的E-Pdoc申請系統,這一系統讓WIPO得以用電子形式接收、處理和發送國際專利優先權文件。有了此一電子申請系統,申請人可以要求同一件申請案以其在任何特定簽約國專利局首次提出申請的日期?國際專利申請日。如果申請得到有關國家專利局的專利授權,該先申請日還可以作?獲得國際專利有效保護的起始日期。 受到電子申請系統方便性之鼓舞, 2004年國際專利申請數量激增並正式突破了一百萬件申請的大關,同一年依據專利合作條約(PCT)規定所提交申請的數量也創下紀錄,共計12萬多件。其中美國繼續列在最大用戶榜首,但增長速度最快的是亞洲大陸─即:日本、韓國和中國大陸。
美國商務部放寬對中國特定晶片出口許可簽審政策美國商務部產業安全局(Bureau of Industry and Security, BIS)於美東時間2026年1月14日公告修正《出口管制規則》(Export Administration Regulations, EAR)對「特定半導體」出口至中國與澳門的許可簽審政策,將原本自2022年10月7日拜登政府時期的「推定拒絕」(presumption of denial)改為「逐案審查」(case-by-case review),並於美東時間2026年1月15日生效。 本次修正將放寬「特定半導體」原先較難輸往中國的法規障礙,影響的貨品範圍包括先進運算類貨品,即總處理效能(Total Processing Performance)低於21,000,且動態隨機存取記憶體(DRAM)總頻寬低於6,500GB/s的先進晶片(NVIDIA H200、AMD MI325X,或與NVIDIA H200同等級貨品);以及較前述晶片規格與效能成熟的貨品。 出口人適用本次修正之「逐案審查」許可簽審政策,須滿足以下條件: 1.欲出口之半導體相關貨品在本次公告時,已可在美國商業市場銷售。 2.欲出口之貨品在美國境內具有充足供應。 3.為銷往中國而生產的晶片,不會排擠全球晶圓代工產能,亦不會影響給美國最終使用人相同或更先進貨品的供應情形。 4.出口貨品須在美國境內接受獨立第三方實驗室檢測(testing),加以確認貨品的技術能力與功能,如同出口人許可申請文件所述。 而在中國的收貨方,須已有充分的貨品安全管控程序。BIS此要求並未同步放寬EAR既有的最終用途、最終使用人查核與流向管理標準。建議出口人於交易前、交易中與交易後,積極與供應鏈夥伴建立良好法遵機制。
WIPO公布《2021年世界智慧財產權指標報告》全球商標註冊申請在疫情影響下仍大幅上升2021年11月8日,世界智慧財產權組織(World Intellectual Property Organization,簡稱WIPO)發布2021年《世界智慧財產權指標報告》(World Intellectual Property Indicators Report,簡稱WIPI)。報告指出全球的商標申請在2020年成長了13.7%、專利成長1.6%、外觀設計成長2%。WIPO執行長表示:「WIPO世界智慧財產權指標報告證實,儘管世界經濟出現數十年來最嚴重的緊縮,但智財權申請——一個強而有力的創新指標——在疫情期間展現出非凡的復原力」。本報告以2020年度,蒐集自世界各地150個官方智財組織、以及WIPO的申請、註冊和延展的統計數據為依據,分析全球智慧財產權活動,範圍涵蓋專利、新型、商標、工業外觀設計、微生物、植物品種保護和地理標誌。 WIPO每年皆會收集和分析官方智財統計數據,發布年度WIPI報告,為政策制定者、商業領袖、投資人、學者和其他欲了解、分析智財生態宏觀趨勢的人提供全球智財資訊。