歐洲央行發表「虛擬貨幣架構」報告,法國比特幣交易平台取得PSP資格。

歐洲央行(European Central Bank,簡稱ECB)於2012年10月29日提出「虛擬貨幣架構(Virtual Currency Schemes)」報告(全文可至歐洲央行網站下載,下載網址: http://www.ecb.int/pub/pdf/other/virtualcurrencyschemes201210en.pdf),Bitcoin(中譯「比特幣」)為該報告的研究重點。該報告將虛擬貨幣架構分為三類:1. 封閉性虛擬貨幣架構,與實體經濟幾乎無連結,通常用於遊戲中,例如暴雪娛樂(Blizzard Entertainment)的魔獸金幣(World of Warcraft Gold, WoW Gold);2.單向貨幣流(通常是流入) 的虛擬貨幣架構,可以現金依照匯率兌為虛擬貨幣後用於購買虛擬商品或服務,少數例外可使用於購買實體商品或服務,這類型代表如臉書的FB幣;3. 雙向貨幣流的虛擬貨幣架構,類如其他一般貨幣,具有買進賣出匯率,可支應虛擬以及實體商品、服務的買賣,如比特幣。基於本身特性,比特幣並非歐盟電子貨幣指令(Electronic Money Directive, 2009/110/EC)以及歐盟支付服務指令(Payment Service Directive,簡稱PSD,Directive 2007/64/EC)的適用範圍。目前虛擬貨幣欠缺與實體經濟的聯結,交易量小且欠缺廣泛的使用接受度,因此對於金融以及物價的穩定影響有限;另外,虛擬貨幣欠缺妥適的法規管制,可能被用於不法活動,如犯罪、洗錢、詐欺等等;綜上,若任由虛擬貨幣持續發展而不管制,將被視為是中央銀行的失職而影響其聲譽。然而,報告指出,基於下述原因,虛擬貨幣將有可能繼續蓬勃發展:1. 網路以及虛擬社群使用人的持續增加;2. 電子商務以及特定數位產品的發展,提供虛擬貨幣架構良好的發展平台;3. 相較於其它電子支付產品,虛擬貨幣具備較佳匿名性;4. 相較於傳統支付工具,虛擬貨幣具備較低交易成本;5. 提供虛擬社群所需要的較直接以及快速的交易清算特性。
比特幣出現於2009年,透過數理運算的「挖礦(mining)」技術產生,無發行人,屬於點對點(peer to peer)虛擬貨幣,可匿名持有交易。目前一枚比特幣約當13塊美金,大約新台幣390元,可用於國際部落格平台Word Press,美國紐約、舊金山的部份實體商店也接受比特幣付費。基於比特幣本身的設計,比特幣的流通數量有限,市面上目前流通的比特幣約有1050萬個,預估至2014年將可全數開鑿完畢。全世界最大的比特幣交易所為東京的Mt.Gox,市占率超過80%,支援美金、英鎊、歐元、加幣、澳幣、日圓以及波蘭幣。2011年時,法國法院在Macaraja v. CIC Bank一案指出,點對點比特幣交換為支付服務,在法國應取得PSP執照。
由法國軟體公司Paymium所建置的比特幣交易平台Bitcoin-Central,於2012年12月與取得PSD支付服務提供者(Payment Service Provider,簡稱PSP)執照的法國業者Aqoba結盟,因而取得PSP資格。依照PSD附件說明,所謂的支付服務,包含存款、提款、轉帳、匯款以及第三方支付服務。透過PSP,Bitcoin-Central在歐盟法制架構下取得與Paypal相同的地位,與銀行業者的重大差別只在於Bitcoin-Central無法提供貸款服務。
Bitcoin-Central提供簡易的比特幣交易界面服務,甚至還有手機錢包(mobile wallet),消費者只消在Bitcoin-Central註冊就可以儲值、購買、交易比特幣並將比特幣轉換為現金,也可以當成薪資帳戶直接存入薪資,未來Bitcoin-Central可發行Debit-Card提供刷卡消費的功能。目前Bitcoin-Central只支援比特幣與歐元的轉換服務,而不提供其他幣別的轉換服務。Bitcoin-Central透過PSP業者Aqoba持有服務使用人儲值之歐元款項,款項存放於法國銀行Credit Mutuel,與Paymium的自有款項切割管理。上述歐元款項受有與一般銀行存款相同的法國中央存款保險"Garantine des dépôts”保障,但是比特幣款項由於並不存放於銀行,因此並不受存款保險保障。
比特幣最常被詬病之處在於其常被用於洗錢以及毒品買賣等等犯罪活動,但是支持者指出,現金不也是有相同問題嗎? 現金的洗錢防制透過金融監理的銀行監管進行,虛擬貨幣之交易平台未來也將是法規管制重點。縱使有部分比特幣支持者反對將比特幣納入法制管理,認為比特幣應該依照其原始設計理念運行,Bitcon-Central與PSP業者Aqoba合作可視為對於ECB之正面回應,為虛擬貨幣法制管理之重要進展。
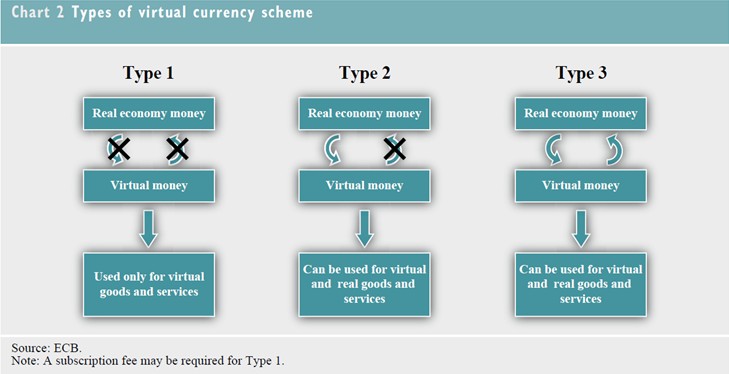
資料來源:European Central Bank, Virtual Currency Schemes, 15 (2012)
- Rosemary Westwood, Why Bitcoin is the banking industry’s newest, biggest threat_
- Bitcoin-Central backtracks on PSP claims,FINEXTRA, Dec. 7, 2012
- Bitcoin digital currency site to operate like bank in France, CBC NEWS, Dec.7, 2012
- Jemima Kiss, Virtual currency Bitcoin registers with European regulators, THE GUARDIAN,Dec.7, 2012
- davout, Bitcoin-Central, first exchange licensed to operate with a bank, This is Huge, BITCOIN FORUM, Dec.6, 2012
- Jon Matonis, ECB: "Roots Of Bitcoin Can Be Found In The Austrian School Of Economics",FORBES, Nov.03, 2012

謝孟珊
資深法律研究員 編譯整理
davout, Bitcoin-Central, first exchange licensed to operate with a bank, This is Huge, BITCOIN FORUM, Dec.6, 2012, https://bitcointalk.org/index.php?topic=129461.0 (last visited Jan. 2, 2013)
Jemima Kiss, Virtual currency Bitcoin registers with European regulators, THE GUARDIAN,Dec.7, 2012, http://www.guardian.co.uk/technology/2012/dec/07/virtual-currency-bitcoin-registers (last visited Jan. 2, 2013)
Bitcoin digital currency site to operate like bank in France, CBC NEWS, Dec.7, 2012, http://www.cbc.ca/news/technology/story/2012/12/07/bitcoins-banking-euro.html (last visited Jan. 2, 2013)
Bitcoin-Central backtracks on PSP claims,FINEXTRA, Dec. 7, 2012, http://www.finextra.com/News/FullStory.aspx?newsitemid=24361 (last visited Jan. 2, 2013)
Rosemary Westwood, Why Bitcoin is the banking industry’s newest, biggest threat_
The cold, hard cash of the Internet has seen dramatic growth recently, and shows no signs of slowing down,MACLEANS.CA, Jan.2,2012, http://www2.macleans.ca/2013/01/02/why-bitcoin-is-the-banking-industrys-newest-biggest-threat/#more-332780 (last visited Jan. 7, 2013)
ECB issues “knee-jerk reaction” analysis of virtual currency risk, E-FINANCE & PAYMENTS LAW & POLICY, Vol.06(11),1,1 (2012).
Bitcoin exchange parterners with PSP for license compliance, E-FINANCE & PAYMENTS LAW & POLICY, Vol.06(12),1,1 (2013).
Jon Matonis, ECB: "Roots Of Bitcoin Can Be Found In The Austrian School Of Economics",FORBES, Nov.03, 2012, http://www.forbes.com/sites/jonmatonis/2012/11/03/ecb-roots-of-bitcoin-can-be-found-in-the-austrian-school-of-economics/ (last visited Jan. 2, 2013)



近期美國與中國大陸雙方針對貿易問題展開激烈攻防,起因為美國冀望透過「貿易戰」扭轉中美龐大的貿易逆差,而其中一個主要爭議點即為中國大陸日趨嚴重之侵權仿冒等問題。 中國大陸於第十三屆全國人大常委會表決通過最高人民法院提請審議的《關於專利等案件訴訟程式若干問題的決定》,批准最高人民法院設立知識產權法庭,主要審理專利等專業技術性較強的知識產權民事及行政上訴案件,以達成知識產權案件審理專門、集中及人員專業化之目的,提供更為專業之司法服務及保障。由最高人民法院知識產權法庭統一審理發明和實用新型專利為主之上訴案件,有利於對中外企業知識產權之保護,實現知識產權效力判斷與侵權判斷兩大訴訟程式和裁判標準的對接,以利解決機制上之裁判尺度不一問題,提高知識產權審判品質效率,提升司法公信力。 值得注意的是,最高人民法院知識產權法庭審理之案件,僅以不服知識產權一審判決、裁定中發明和實用新型專利等案件,其他上訴案件,仍由智慧財產權法院所在地的高級人民法院審理。中國大陸最高人民法院院長周強表示,知識產權法庭之設立,宣示平等保護中外市場主體知識產權,該知識產權法庭並不會處理與不正當競爭、商標或營業秘密有關之案件。
美國資訊安全分析新挑戰:巨量資料(Big Data)之應用在2013年的國際資訊安全會議(RSA Conference)上,資安專家紛紛表示,將Big Data技術應用於資訊安全分析的項目上,確實可以幫助企業建立更佳的情勢判斷能力,但在實際執行過程中是一大挑戰。 資安廠商如RSA和賽門鐵克公司,在會議上表示目前的策略是透過新的數據匯集、比對和分析協助企業篩選、過濾結構化和未結構化資料的威脅指標,這是傳統的特徵偵測(signature-based)安全工具無法做到的。 不像傳統的安全手段著重於阻斷攻擊,新的技術強調偵測並立即回應違犯行為,也就是提前遏止任何違犯行為,協助企業作全面性的偵測而不擔心有所遺漏。 由於越來越多的美國政府機關和民間企業遭受到針對性和持續性的攻擊,巨量資料技術的應用需求激增。企業內部都累積著大量的數據和多元的數據種類,而需要動新技術來保護這些數據資料免於惡意人士或對手的竊取或其他侵害行為。企業應該要因應實際面臨的威脅和所獲悉的威脅情報來建立安全模型,取代部署特定產品和外圍系統的防禦。 美國無論是政府機關或民間企業都被捲入了不對稱戰爭-對手是武器精良、準備充分並有嚴密組織的網路敵人。 「駭客只需要攻擊成功一次,但我們必須每次都是成功的」賽門鐵克的總裁deSouza表示。「因此與其專注的在阻擋所有威脅,更好的辦法是使用巨量資料技術偵測侵入行為並消解之」。而在會議中資安專家都肯認至少從理論上來說,以巨量資料技術強化資訊安全是很好的想法。 不過另有其他的說法,金融服務企業LSQ的首席安全及法務主管皮爾遜認為,許多人的電腦紀錄檔和所有的電子裝置都早就被侵入滲透了,這才是問題所在。他表示,目前現存的SIEM(安全性資訊及事件管理)工具可以讓企業聚集來自許多個安全設備的巨量登錄數據整合在同一系統內,但真正的問題是,SIEM工具必須要有能力分析數據並找出關聯性,如此才能偵測到駭客入侵的前兆證據和真實的入侵行為,這和彙整數據是不同的兩件事。許多企業所面臨的問題不是缺乏數據資料,而是要如何為資訊安全的目的建立關聯規則和應用方式,以有效率的方式找出有用的巨量數據並進行分析,和留下可供進行訴訟使用的證據。
英國BEIS發布第一代(SMETS1)智慧電表相容性公眾諮詢英國商業、能源和產業策略部(Business, Energy and Industrial Strategy, BEIS)於2018年4月17日發布公眾諮詢,議題為「最大化第一代(SMETS1)智慧電表的相容性(interoperability)」,該諮詢將截止於2018年5月24日。 英國對於SMETS1的推廣分為兩階段進行,基礎建設階段始於2011年,主要安裝階段則於2016年11月開始,國家數據及通訊供應商-資料通訊公司(Data Communications Company, DCC)自此階段開始營運,直至2020年智慧電表建置完成。 因現今由各能源供應商使用自身資料及通訊設備裝設第一代智慧型電表,造成消費者無法任意更換能源供應商之情況。對此,英國政府之長期政策目標雖為SMETS1最終可全數透過DCC進行運作,然由於現階段尚未強制能源供應商使用DCC所提供之服務,使用SMETS1的消費者仍無法自由的轉換能源供應商。 本文件提出了兩個方案向公眾諮詢: 要求能源供應商於六個月時限內至DCC註冊其所提供且合於規範的SMETS1,或將SMETS1更換為SMETS2(第二代智慧電表)。而於2020年12月31日前,所有未註冊之SMETS1將強制更換為SMETS2。 若能源供應商已嘗試所有合適的解決方法,仍無法於2019年底前使SMETS1在智慧模式下運作,就必須在2020年6月底前將SMETS1更換為SMETS2。 若供應商係於2019年後才取得SMETS1,於獲得SMETS1之後的六個月內採取所有相關措施後仍無法令SMETS1以智慧模式運作,亦應更換為SMETS2。最終,所有不能運行智慧模式之SMETS1將於2020年12月31日前被完全汰換。 英國政府期透過更完善的政策規劃改善現階段SMETS1透過個別能源供應商之數據及通訊系統運作之情況,以確保SMETS1之智慧模式於消費者更換供應商時能維持正常運作,使消費者可確實獲取改用智慧電表之利益。我國於2015年已開始推動低壓智慧電表建置,英國面臨之問題值得借鏡,政府於推廣低壓智慧電表之同時應注意智慧電表基礎設施之相容性,以增進低壓智慧電表建置效率及降低建置成本。
美國FDA於20250617宣布將試行「局長國家優先審查券」COVID-19疫情後美國開始積極處理藥品供應鏈脆弱性,為提振本土製造與審查效率,美國食品及藥物管理局(Food and Drug Administration, FDA)於2025年6月17日宣布將試辦「局長國家優先審查券」(Commissioner’s National Priority Voucher, CNPV)。該計畫依據《聯邦食品、藥品與化妝品法》(The Federal Food, Drug, and Cosmetic Act, FFDCA)與《公共衛生服務法案》(Public Health Service Act, PHSA)授權。CNPV將不同審查分組集中處理,並結合資料預先提交機制,力求將一般10-12個月的審查流程壓縮至1-2個月,試辦期為一年,並與現行優先審查及優先審查券(Priority Voucher, PRV)機制獨立並行。 內容要點: 1.遴選資格:符合任一「國家優先」標準之廠商 因應公衛危機:如廣效疫苗開發 帶來潛在的創新療法:超越突破性療法認定成效的新型療法 解決未滿足公共衛生需求:如罕病或缺乏療效標準治療之疾病 提升美國供應鏈韌性:如將藥品研發、臨床、生產遷至美國 提高可負擔性:將美國藥價降至最惠國藥價,或減少下游醫療費用 2.使用與要求: 適用階段:可於申請臨床試驗或申請藥證等階段啟用,亦可先領「未指名券」保留資格。 文件要求:需提前60天提交完整藥品化學製造與管制(Chemistry, Manufacturing, and Controls, CMC)與仿單預審,如遇重大缺件FDA得延長審查期限。 有效性:2年內使用,逾期失效;不可轉讓,但併購案中可沿用。 CNPV透過團隊同日決策,有望在FDA人力縮減背景下縮短審查時程。並強調國家利益,可能優先惠及具戰略價值及在美投資的大型藥廠;對我國優化藥品審查流程與吸引製造投資等目標,亦具重要參考價值。