新德國包裝法簡介

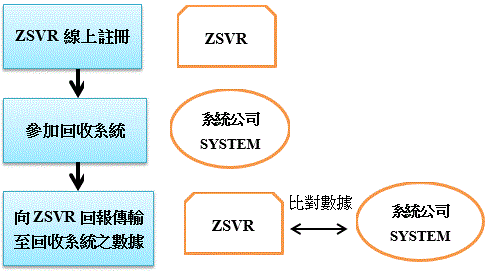
為有效降低包裝廢棄物對環境造成的汙染及不利影響,使製造商履行其B2C(business to customer)產品責任,德國以新的包裝法(Packaging Act, VerpackG)取代現行的規範(Packaging Ordinance,VerpackV),並已於2019年1月1日生效。
新包裝法VerpackG不同於VerpackV之處,在於除要求業者須加入原有的回收系統外,另授權Zentrale Stelle(Stiftung Zentrale Stelle Verpackungsregister,ZSVR)基金會作為新包裝法強制登記制度的執行單位,規範欲在德國銷售產品包裝之所有實體或網路製造商及零售商,有義務於ZSVR的數據資料庫”LUCID”註冊,才能在德國地區進行銷售,並且為全面提升透明度,乃規範於LUCID註冊之商家資訊皆屬可供大眾公開查詢。
依VerpackG規定,於2019年1月1日起未為註冊的商家,其包裝商品不能在德國上市,否則恐將臨100,000歐元之罰款;另未加入回收系統之商家,恐面臨200,000歐元之罰款。而除須註冊與回收系統的加入外,製造商及零售商尚須將以下之包裝相關資訊提供給ZSVR做比對:
(一)註冊號碼(商家於資料庫註冊時,由ZSVR所提供之註冊號碼)
(二)包裝材料及容積
(三)製造商履行生產者延伸責任(Extended Producer Responsibility)簽訂的包裝方案名稱
(四)與回收公司或回收系統間簽訂之契約期限
資料來源:自行繪製
圖 德國包裝法實施步驟

蔡宛君
法律研究員 編譯整理
How-To Guide to the Packaging Act for Manufacturers, Stiftung Zentrale Stelle Verpackungsregister(2018), https://www.verpackungsregister.org/fileadmin/user_upload/How-to-Guide_en_13072018_final.pdf (last visited Jun. 1, 2019)
The ten most important questions regarding the implementation of the Packaging Act ,Stiftung Zentrale Stelle Verpackungsregister(2018), https://www.verpackungsregister.org/fileadmin/user_upload/10-W-Fragen_en_13072018_final.pdf (last visited Jun. 1, 2019)
The new German Packaging Act is here – and it’s particularly important for online retailers,Der Grüne Punkt, https://www.gruener-punkt.de/en/services/packaging/german-packaging-act.html (last visited Jun. 4, 2019)



為加強兒少網路安全,英國通訊局(Office of Communication)於2025年4月24日發布「兒少風險評估指導原則」(Children's Risk Assessment Guidance)以及「兒少保護行為準則」(Protection of Children Codes of Practice),以供受規範之網路服務提供者遵循。 「兒少風險評估指導原則」要求所有受《網路安全法》(Online Safety Act 2023)規範的「使用者對使用者服務」(user-to-user service)和「搜尋服務」(search service)的服務提供者,在完成兒少存取評估(children’s access assessments)確認服務有可能被兒少存取後,必須在三個月內即2025年7月24日前,完成兒少風險評估(children’s risk assessments)。 至於「兒少保護行為準則」一經國會審議通過,則自2025年7月25日起生效,服務提供者必須採取「兒少保護行為準則」規定的安全措施(safety measures),或實施其他有效手段,以減少上開風險發生的可能性。 「兒少保護行為準則」列出的安全措施包括: 一、更安全的資訊發送: 有運作推薦系統(recommender system)且存在中度或高度有害內容風險的服務提供者,應調整其演算法,確保不向兒少推播有害內容。 二、有效的年齡驗證: 風險最高的服務提供者必須使用高度有效的年齡驗證,以識別使用者是否為兒少。 三、有效的內容審查: 服務提供者應建立內容審查機制,一旦發現有害兒少的內容時,能迅速採取行動。 四、更多的選擇與支援: 服務提供者應賦予兒少更多的控制權,包括允許對內容或貼文表達不喜歡、接受或拒絕群組聊天邀請、封鎖用戶、將用戶設為靜音、以及關閉自己貼文的留言功能等。 五、簡便的舉報與申訴: 服務提供者應提供透明的舉報與申訴管道,且確保提出舉報和申訴的管道和方式對兒少而言相對輕鬆且簡單。 六、強化的治理: 服務提供者應設置負責兒童安全的專責人員,高層團隊並應每年審查兒童風險管理情況。 如服務提供者未能履行上開義務,英國通訊局可處以罰款。必要時,甚至可向法院聲請禁制令封鎖該平台。 處在網路無限擴張的時代,保障兒少網路安全絕對是重要且不容忽視的課題。英國通訊管理局發布的實施作法,非常值得我國觀察學習與參考。
我國生物與遺傳資源權利歸屬及管理思維初探 英國同意BT「下世代文字中際服務申請」為了讓聽力或語言障礙之民眾,取得更為便利的電信服務,英國Ofcom在2012年10月透過「回顧中繼服務-決策下世代文字中繼服務」(Review of relay services: Decision on the introduction of Next Generation Text Relay)陳述書(Statement)之發佈,提高通訊服務業者 (communications providers)對身障者的義務。根據陳述書內容,英國市話、行動通信業者必須在2014年4月18日,達成「下世代文字中繼服務」(Next Generation Text Relay,NGTR)之要求。因此,英國電信BT於去(2013)年11月提出審查申請,並在今年3月獲得Ofcom許可。 Ofcom要求下世代中繼服務最主要重點,主要可分為(1)透過網際網路的連接,提供雙向語音服務,讓雙方談話更為流暢;(2)促使更多載具皆可使用文字中繼服務。BT為了落實上述核心要求,除了透過提升服務性能、技術應變能力、員工培訓與申訴機制,增加中繼服務的品質,以通過Ofcom核可外,BT亦允諾2014年4月18日後,該公司文字中繼服務將可進一步延伸至: 1.既有文字電話(textphones)與視障閱讀器(Braille readers)。 2.支援Windows XP系統以上個人、筆記型電腦。 3.適用Linux的Intel個人、筆記型電腦。 4.Intel型Mac OS X版本。 5.行動電話具有Android 4的作業系統。 6.2014年6月30日將可提供服務至蘋果公司相關產品,包括iPhone與iPad。 BT不僅提供文字中繼服務於自身客戶,亦提供批發接取(Wholesale access)於其他通訊服務業者,使其具備文字中繼服務,讓非BT的消費者亦可獲得無障礙服務。相信隨著更多通訊服務業者提升文字中繼服務功能後,將可讓聽障、語言障礙民眾使用電信服務更加便利,使英國充分落實聯合國「身心障礙者權利公約」之精神。
澳洲法院判決BRCA1基因專利部分無效澳洲高等法院(澳洲的最高司法機關)在10月7日時做出重要判決,認為單純從人類基因體分離出來的基因序列,不足以作為專利的申請標的。本案的原告是一名69歲曾罹患乳癌的女士,他向法院起訴請求法院宣告Myriad 基因公司所擁有的BRCA1基因專利中部分的專利範圍(claim)無效。BRCA1基因的突變(mutation)或特定的表型被認為與乳癌及卵巢癌的發生機率有關。在先前的審級,法院都判決被告勝訴,但高等法院推翻了先前的見解,以7票贊成0票反對的比數\判決原告的上訴有理由,Myriad基因公司的專利無效。本案由首席法官連同其餘三位法官提出多數意見,另外有兩份協同意見。 本案的主要爭點在於系爭專利是否符合澳洲1990年專利法(Patents Act 1990)中,對專利應屬於一種「生產方式」(manner of manufacture)的規定。多數意見認為系爭的專利範圍只是BRCA1基因的序列,這些資訊並非由人類所「製造」,而僅是由人類所辨別。因此這無法被視為是一種生產的方式,不符合專利法的相關要求。若要將其視為生產方式,則需要進一步擴張生產方式的概念範圍,不適合由法院進行判斷。同時法院認為這個專利可能造成寒蟬效應,使得與BRCA1相關的分離技術變得過於昂貴或形成事實上的壟斷狀態,也與專利法希望促進發明的初衷不符。最後,法院在確認澳洲對於是否應承認此類專利並無國際法上的義務後,宣告此專利無效。 澳洲的學界對此判決表示歡迎,認為此判決將使醫療人員執行職務時免於侵犯智慧財產權的恐懼。但澳洲的生技產業則認為這個判決可能會打擊相關的研究,造成負面影響。澳洲法院的判決與美國先前的判決見解相當類似,同時該判決也可能對於其他國家的類似案件發生影響。例如在加拿大的一個與罕見心臟疾病基因有關的官司,就很可能會受到本判決的影響也宣布該基因專利無效。