新加坡資料共享法制環境建構簡介

新加坡資料共享法制環境建構簡介
資訊工業策進會科技法律研究所
2019年12月31日
壹、事件摘要
如何有效運用資料創造最大效益為數位經濟(Digital Economy)重點,其中資料共享(data sharing)是有效方法之一。新加坡自2018年以來推動「資料共享安排」機制(Data Sharing Arrangements, 下稱DSAs)與「可信任資料共享框架」(Trusted Data Sharing Framework),建構資料共享環境,帶動國內組織[1]資料經濟發展與競爭力。
貳、重點說明
自從2014年新加坡政府推行「2025智慧國家(Smart Nation)」以來,即積極鋪設國家數位經濟建設,大數據資料分析等數位科技發展為其重點,預估2022年60%國內生產總值將與數位經濟有關[2] 。其中,希望透過資料共享促進組織、政府、個人三方間資料無障礙流通,降低蒐集、處理與利用成本,創造更多合作機會進行創新應用,因此從法制面、環境面與技術應用層面打造完善的資料共享生態系統(data sharing ecosystem)[3]。
然而依據《個人資料保護法》(Personal Data Protection Act 2012,下稱個資法)第14條以下規定,組織蒐集、處理與利用個人資料應取得當事人同意,除非符合第17條研究目的等例外情形。由於資料共享強調可將資料進行多節點快速傳遞近用,使資料利用價值最大化,因此若依據個資法規定每次共享皆須事前獲得當事人同意,將使近用成本增高並間接造成資料流通產生障礙。因此為因應國家政策與產業需求,新加坡個人資料保護委員會(Personal Data Protection Commission, 下稱個資委員會)依據個資法第62條所賦予的豁免權(exemption),個人或組織可在遵循個資委員會訂定的規則下,依照個案給予組織免除個資法部分規範[4] ,而DSAs機制即是一種[5]。
DSAs是由個資委員會於2018年設立的沙盒(sandbox)計畫,如組織所進行的共享模式是在特定群體並範圍具體明確,同時不會造成個人有負面影響等情事,可在不須經個人同意下進行資料共享[6]。並且,為進一步提升組織與消費者間信任,2019年6月個資委員會與資訊通信媒體發展局(Info-Communication Media Development Authority of Singapore,下稱資通發展局)共同推出「可信任資料共享框架」指南建議,由政府擔任監管角色,組織只要符合指南建議方向,如遵循法律、達到一定資料技術應用品質與實施資安與個資保護措施下,可以進行個人與商業資料之共享,DSAs機制是共享方法之一。以下簡述新加坡個資法規範、指南建議與DSAs機制運作方式。
圖1:資料共享環境建構
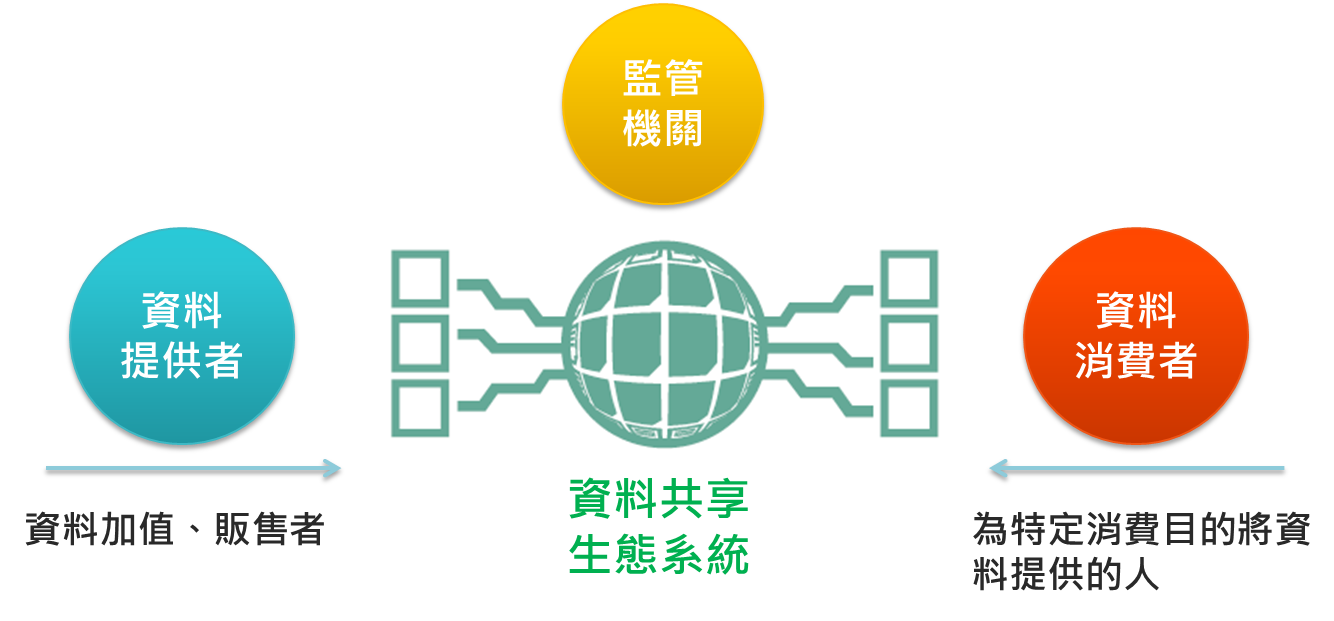
資料來源:新加坡資通發展局
一、新加坡個人資料保護法規範
在沒有個資法第17條所列之例外情形下,依據第14條以下規定,組織如近用個人資料應獲得個人同意,同時應符合目的使用及通知義務,尤其應給予個人可隨時撤回同意之權利[7]。
同時組織應根據個人要求,提供近用個人資料之方法、範圍與內容,以及更正錯誤資料權利[8]。並且組織必須任命資料保護官(Data Protection Officer, DPO)隨時向大眾提供通暢的個資聯絡管道,來確保個資透明性與完整性[9]。
在資料保護措施上應有合理安全的資安防護技術,以保障資料不被未經授權近用的風險。當使用目的不在時,需妥善保留或予以去識別化,同時如須境外轉移資料時,境外之資料保護措施應至少與新加坡個資法規範標準相同[10]。
二、免除同意之DSAs機制
DSAs機制是由個資委員會於2018年設立的沙盒(sandbox)計畫,也就是組織可透過申請免除資料共享前必須獲得個人同意之規範。然而如組織擬向個資委員會申請DSAs機制,必須符合三個條件[11]:
- 共享範圍需在特定群體、期間與組織內:即只限定在具體特定的應用情境內,若超出申請範圍,例如分享至其他非申請範圍的組織,則須再經過個資委員會批准[12]。
- 近用目的需具體明確:即資料共享必須應用於特定且明確目的,如以「社會研究目的」作為申請則範圍過大不夠明確[13]。
- 近用資料對於個人不會有不利影響,或公共利益大於個人利益:例如共享目的不是直接用於銷售或存在合法利益,或是共享本身具備公共利益且明顯大於個人可預見的(foreseeable)不利影響,此時個資委員會可考慮同意組織申請免除[14]。
三、建立以信任為基礎之資料共享模式
雖然取得DSAs機制免除同意可以使資料近用方式更為簡便,然而在進行資料共享前,仍應有完善的技術品質與資安保護措施,因此在「可信任資料共享框架」指南建議中,組織應透過法律遵循、導入AI或區塊鏈等新興技術,並具備相應資安保護措施來建構可信任的資料共享環境,實際步驟可分為以下四階段[15]:
圖2:可信任資料框架
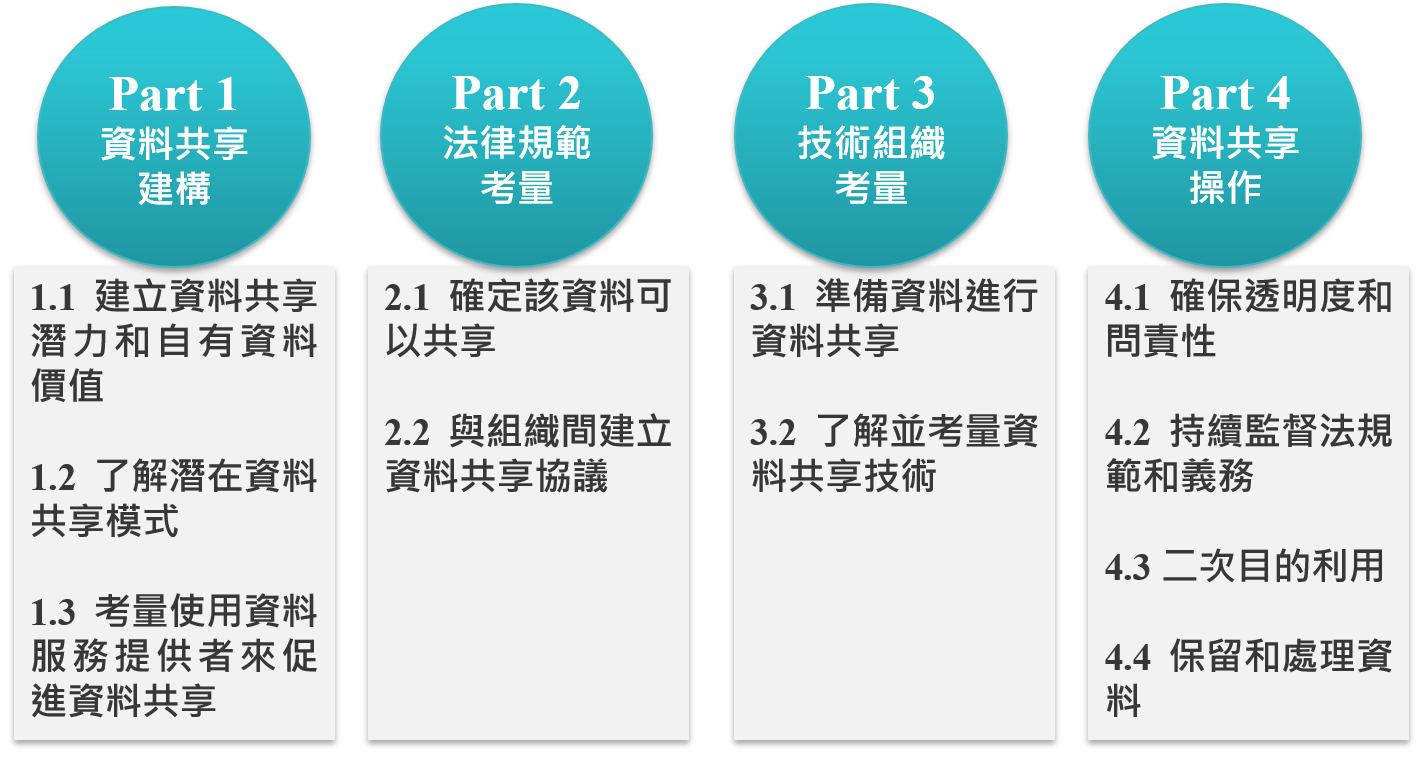
資料來源:新加坡資通發展局
第一階段為「資料共享建構」[16],由組織自行評估存有的商業或個人資料是否具共享價值與潛在利益,並要如何進行共享,例如資料共享方式屬於雙邊(bilateral)、多邊(multilateral)或是分散式(decentralized,又稱「去中心化」)。以及資料種類有哪些,如主資料(master data)、交易資料、元資料(metadata)、非結構化資料(unstructured data)等。組織可將資料共享方式、種類依據無形資產(intangible asset)評價方式,即市場法(market approach)、成本法(cost approach)與收入法(income approach)三種評價方法進行評價,來衡量共享之價值性。除資料價值判斷外,組織必須自行評估自身組織與將來之合作夥伴是否有足夠能力管控共享之資料,包括是否具備一定技術能力的資安與資料保護措施等。
第二階段為「法律規範考量」[17],即決定哪些資料可以進行共享,從規範面檢視個資法、競爭法與銀行法等是否有例外不得共享規定,例如信用卡號碼或個人生物識別資訊不得共享。若資料共享類型不會對個人造成不利影響或具備公共利益,並有通知(notification)個人給予選擇退出(opt-out)的機會,組織可依個案申請DSAs機制之豁免。同時另外鼓勵組織向IMDA申請資料保護信任標章(Data Protection Trustmark, DPTM)認證,透過認證機制使消費者更能信任組織運用其個人資料[18]。
第三階段為「技術組織考量」[19],包含組織是否有能力建立資安風險管理與個資侵害之因應措施,是否有即時將資料安全備份技術,並針對不同傳輸技術如有線/無線網路、遠端存取(VPN)、應用程式介面(API)、區塊鏈等區分不同資安防護與風險管理能力。
最後一階段為「資料共享操作」,當已準備進行資料共享時,需再次檢視是否已符合前三個階段,包含透明性、責任義務、法律遵循、近用資料方式與取得目的外利用同意等[20]。
參、事件評析
個人資料視為21世紀驅動創新的重要價值,我國部會亦開始討論「個資資產化」的可能[21]。面對數位經濟時代來臨,有效運用數位科技將潛藏個人資料的大數據進行加值利用,不僅有利組織與創新發展,更可回饋消費者享有更好的產品與服務。
新加坡政府以資料共享作為數位經濟發展重點方向之一,在具備一定程度技術能力、資安保護措施與組織控管之條件下,可向主管機關申請免除個人同意之規範。透過一定法規鬆綁讓資料利用最大化以創造產業創新價值,同時依據主管機關要求的保護措施,使消費者信賴個人資料不會遭受不當利用或侵害。DSAs機制與「可信任資料共享框架」指南之建立,適時調適個人資料保護規範與資料應用間的衝突,並提供組織進行資料共享之依循建議,作為推動該國數位經濟發展方針之一。
[1]組織(organisation)依據新加坡個人資料保護法(Personal Data Protection Act 2012)第2條泛指個人、公司、協會、法人或團體。
[2]INFOCOMM MEDIA DEVELOPMENT AUTHORITY 【IMDA】, Trusted data sharing framework (2019), at 7, https://www.imda.gov.sg/-/media/Imda/Files/Programme/AI-Data-Innovation/Trusted-Data-Sharing-Framework.pdf (last visited Sep. 11, 2019).
[3]id.
[4]Personal Data Protection Act 2012 (No. 26 of 2012) §62, “The Commission may, with the approval of the Minister, by order published in the Gazette, exempt any person or organisation or any class of persons or organisations from all or any of the provisions of this Act, subject to such terms or conditions as may be specified in the order.”
[5]Data Sharing Arrangements, PDPC, https://www.pdpc.gov.sg/Overview-of-PDPA/The-Legislation/Exemption-Requests/Data-Sharing-Arrangements (last visited Dec. 1, 2019).
[6]id.
[7]IMDA, supra note 2, at 31; Personal Data Protection Act 2012 (No. 26 of 2012) §14, 16, 20.
[8]id. Personal Data Protection Act 2012 (No. 26 of 2012) §21.
[9]IMDA, supra note 2, at 31.
[10]id. at 32. Personal Data Protection Act 2012 (No. 26 of 2012) §24-26.
[11]id.
[12]PERSONAL DATA PROTECTION COMMISSION【PDPC】, Guide to Data Sharing (2018), at 14, https://www.pdpc.gov.sg/-/media/Files/PDPC/PDF-Files/Other-Guides/Guide-to-Data-Sharing-revised-26-Feb-2018.pdf (last revised Oct. 3, 2019).
[13]id.
[14]id.
[15]PDPC, supra note 4. at 28.
[16]id. at 21, 23-25.
[17]id. at 35
[18]id. at 30. Data Protection Trustmark Certification, IMDA, https://www.imda.gov.sg/programme-listing/data-protection-trustmark-certification (last visited Sep. 26, 2019).
[19]id. at 41-47.
[20]id. at 50-51.
[21]林于蘅,〈自己的個資自己賣!國發會擬推「個資資產化」〉,聯合新聞網,2019/06/17,https://udn.com/news/story/7238/3877400 (最後瀏覽日:2019/10/1)。
- 【台北場1】通訊傳播事業個資法遵教育訓練
- 【台中場】通訊傳播事業個資法遵教育訓練
- 【新北場】通訊傳播事業個資法遵教育訓練
- 【高雄場】通訊傳播事業個資法遵教育訓練
- 【台北場2】通訊傳播事業個資法遵教育訓練
- 零售業個資法遵實務與資訊安全說明會
- 114年資訊服務業者個資安維辦法宣導說明會
- 【線上】資訊服務業個資安維計畫說明會
- 【實體】資訊服務業個資安維計畫說明會暨交流工作坊
- 零售業個資法遵實務與資訊安全說明會(台中場)
- 【台北場1】通訊傳播事業個資保護實務專題講座
- 【台北場2】通訊傳播事業個資保護實務專題講座
- 【台北場3】通訊傳播事業個資保護實務專題講座
- 金融相關資服業者線上個資安維宣導說明會
- 【上午場】探索科技法律最前線:資策會科法所「114年成果」分享會暨簡報票選
- 【下午場】探索科技法律最前線:資策會科法所「114年成果」分享會暨簡報票選
- 商業服務業個資管理與資訊安全實務宣導說明會(高雄場)
- 商業服務業個資管理與資訊安全實務宣導說明會(台南場)
- 商業服務業個資管理與資訊安全實務宣導說明會(台中場)
- 商業服務業個資管理與資訊安全實務宣導說明會(台北場)※如遇颱風假延期至7/16
- 【北部場1】國家通訊傳播委員會與通訊傳播產業個人資料保護與管理制度實務工作坊
- 【北部場2】國家通訊傳播委員會與通訊傳播產業個人資料保護與管理制度實務工作坊
- 【北部場3】國家通訊傳播委員會與通訊傳播產業個人資料保護與管理制度實務工作坊
- 【北部場4】國家通訊傳播委員會與通訊傳播產業個人資料保護與管理制度實務工作坊
- 【南部場】國家通訊傳播委員會與通訊傳播產業個人資料保護與管理制度實務工作坊
- 【中部場】國家通訊傳播委員會與通訊傳播產業個人資料保護與管理制度實務工作坊

吳昌鴻
法律研究員 編譯整理



英國商業、能源和產業策略部(Business, Energy and Industrial Strategy, BEIS)於2018年4月17日發布公眾諮詢,議題為「最大化第一代(SMETS1)智慧電表的相容性(interoperability)」,該諮詢將截止於2018年5月24日。 英國對於SMETS1的推廣分為兩階段進行,基礎建設階段始於2011年,主要安裝階段則於2016年11月開始,國家數據及通訊供應商-資料通訊公司(Data Communications Company, DCC)自此階段開始營運,直至2020年智慧電表建置完成。 因現今由各能源供應商使用自身資料及通訊設備裝設第一代智慧型電表,造成消費者無法任意更換能源供應商之情況。對此,英國政府之長期政策目標雖為SMETS1最終可全數透過DCC進行運作,然由於現階段尚未強制能源供應商使用DCC所提供之服務,使用SMETS1的消費者仍無法自由的轉換能源供應商。 本文件提出了兩個方案向公眾諮詢: 要求能源供應商於六個月時限內至DCC註冊其所提供且合於規範的SMETS1,或將SMETS1更換為SMETS2(第二代智慧電表)。而於2020年12月31日前,所有未註冊之SMETS1將強制更換為SMETS2。 若能源供應商已嘗試所有合適的解決方法,仍無法於2019年底前使SMETS1在智慧模式下運作,就必須在2020年6月底前將SMETS1更換為SMETS2。 若供應商係於2019年後才取得SMETS1,於獲得SMETS1之後的六個月內採取所有相關措施後仍無法令SMETS1以智慧模式運作,亦應更換為SMETS2。最終,所有不能運行智慧模式之SMETS1將於2020年12月31日前被完全汰換。 英國政府期透過更完善的政策規劃改善現階段SMETS1透過個別能源供應商之數據及通訊系統運作之情況,以確保SMETS1之智慧模式於消費者更換供應商時能維持正常運作,使消費者可確實獲取改用智慧電表之利益。我國於2015年已開始推動低壓智慧電表建置,英國面臨之問題值得借鏡,政府於推廣低壓智慧電表之同時應注意智慧電表基礎設施之相容性,以增進低壓智慧電表建置效率及降低建置成本。
吃的安心 基改農產品安全性測試系統上路自從1994年第一種基因改造(Genetically Modified , GM)農產品~番茄在美國上市後,越來越多的GM農產品進入了我們的生活,使得大家越來越注重食用的安全性。行政院農業委員會農業藥物毒物試驗所開發的基因改造農產品安全測試系統於11月正式上路,日後台灣自行研發的GM農產品上市前,可以送到藥毒所檢驗,以確定對人體無害。 國際間對於GM農產品安全性爭議主要有兩個層面:生物安全性(作為食品之安全性)與生態環境安全(對環境的衝擊評估)。整體而言,GM農產品的食用安全評估以過敏性測試最為重要,也就是針對轉殖的DNA基因,測試其外源表現物質(蛋白質)對人體的影響,換句話說:蛋白質是較容易讓人體產生過敏的來源。 藥毒所開發的過敏反應和安全性測試系統,其針對GM農產品的評估方法有三:序列比對(和已知過敏原比對)、消化穩定性(採用人工胃液和腸液分解測試)、動物實驗模式(讓大白鼠直接食用)。相信這套安全測試系統的上路,可讓民眾食用台灣自行研發的GM農產品較為安心。
日本公布《空中移動革命藍圖》日本經濟產業省與國土交通省共同組成的「空中移動革命之官民協議會」(空の移動革命に向けた官民協議会),於2018年12月20日第4次會議中公布《空中移動革命藍圖》(空の移動革命に向けたロードマップ,以下簡稱「本藍圖」),期待飛天車(electric vertical take-off and landing, eVTOL)的實現可在都市交通阻塞時或欲前往離島、山間地區等情形下,提供新移動方式,也可運用於災害時的急救搬運及迅速運送物資等。 本藍圖之「飛天車」係電動垂直起降型的自動駕駛航空機,外型近似直升機,並規劃三條發展路線:實際應用目標、制度及標準之整備、機體及技術之研發。從實際應用目標出發,本藍圖規劃自2019年開始進行飛行測試和實證實驗,以2023年投入運用為目標。首先從運送「物品」開始進展到「部分地區的乘客」,2030年代將再進一步擴大實用到「都市中的乘客」。也可應用於災害應變、急救、娛樂等方面。 為了實現上述目標,即需整備機體安全性、技能證明等及未來投入商業應用時所需之各項標準及制度。當然機體及技術之研發也相當重要,透過試作機研發確保並證明機體安全性及可靠性、自動飛行之機上及地面管理系統、確保達到商業化程度的飛航距離及靜肅性之技術。並設定於投入應用後的2025年開始,重新檢討制度及提升技術。
歐盟有條件批准Meta對Kustomer的併購案歐盟執委會於2022年1月27日宣布批准Meta(原Facebook)對Kustomer的併購案。Kustomer公司本身為向企業銷售客戶關係管理(customer relation management, CRM)整合軟體之公司,故本項併購可能影響Meta對消費者資訊的掌握能力,進而提升廣告市場影響力。因之,歐盟執委會基於自去(2021)年8月起深入調查本併購案有無影響公平競爭的結果,作成批准本併購案之決定,但要求Meta應遵守其提出的條件。 依據歐盟執委會的調查結果,主要擔憂本併購案可能阻礙CRM整合軟體之供給市場、以及CRM整合軟體售後客戶服務之供給市場的公平競爭。同時,調查中亦確認到Meta限制Kustomer公司的潛在競爭對手、以及新參與上述市場的業者近用Meta的訊息傳遞路徑應用程式介面(message channel API)。上述潛在競爭對手與業者和Kustomer公司相同,以中小企業為其主要銷售客群。而Meta採取此種經營方式,則可能會劇烈減少CRM整合軟體供給市場、以及CRM整合軟體售後客戶服務供給市場的競爭,導致相關軟體產品或服務的價格上揚,並伴隨品質與創新能量的下降,更可能將之轉嫁予消費者。 對上述調查結果所提出違反公平競爭秩序的疑慮,Meta則提案追加以下約款,作為條件以圖本併購案能夠獲准:(1)Meta保證於10年內,將其訊息傳遞路徑應用程式介面以無償、非歧視的方式,公開予存在競爭關係之客戶服務CRM整合軟體供應商、與新參與市場的業者取用;(2)Kustomer公司之客戶所使用Messenger、Instagram之私人通訊服務,以及WhatsApp之功能未來有進行改良或更新時,Kustomer公司之競爭對手與新進業者同樣得使用該些更新的功能。歐盟執委會最終認為Meta若踐行上述約款,將能消除其違反公平競爭秩序的疑慮,而以Meta履行該些約款為條件批准本併購案。