歐盟「藥品法規包裹法案」(Pharma Package)重點說明與簡析

歐盟「藥品法規包裹法案」(Pharma Package)重點說明與簡析
資訊工業策進會科技法律研究所
2025年09月10日
歐盟理事會(the Council of the European Union)於2025年6月4日就歐盟「藥品法規包裹法案」(Pharma Package)取得一致立場,且正與歐洲議會及歐盟執委會展開三方協商(trilogue),以共同研議最終立法版本[1]。法案預計於2026年初進行表決,若順利通過且加上過渡期,最快將於2028年正式施行,並可能重塑歐盟藥品市場之上市與投資規劃。
壹、背景摘要
有鑑於歐盟現行的藥品法規制度,自2004年修正後施行至今已逾20年,難以充分回應公共衛生需求、藥品可及性落差以及國際市場競爭等多重挑戰。因此,歐盟執委會(European Commission, EC)於2023年4月公布「藥品法規包裹法案」,旨在強化藥品供應鏈穩定性、提升藥品創新與競爭力,以建立更公平、具韌性與永續性的歐洲醫藥市場[2]。而2024年4月歐洲議會(European Parliament)[3]、2025年6月歐盟理事會亦分別提出不同版本,以促進歐洲整合為單一藥品市場。
本次改革對生技製藥產業影響深遠,為協助我國製藥產業在進入歐洲市場前先行掌握相關動向,以下將進行重點說明與分析。
貳、立法重點與比較
一、藥品監管保護期間之修正
在本次藥品法規包裹法案中,有關藥品監管保護期間之修正,為歐盟執委會、歐洲議會與歐盟理事會分歧最大,也是外界高度關注之核心重點。首先,現行歐盟新藥保護是由8年資料專屬保護期(Regulatory Data Protection, RDP)[4]與2年市場獨占期(Market Exclusivity, ME)所構成[5],若於資料專屬保護期間內取得新適應症可再延長1年市場獨占期,亦即保護期共為10~11年。
歐盟執委會則提議將資料專屬保護期縮短至6年,並搭配延長誘因機制,最多可再延長3年,條件包含:
1.滿足現有醫療需求可延長0.5年。
2.藥品在所有歐盟成員國上市可延長2年。
3.進行比較性臨床試驗可延長0.5年。
此外,不同於現行制度將新適應症延長1年納入市場獨占期,歐盟執委會則提議將此移至資料專屬保護期,因此合併原有2年市場獨占期後,保護期共為8~12年。
反之,歐洲議會則擬將資料專屬保護期調整為7.5年,並同樣設有延長誘因機制,但總延長上限至多1年,條件包含:
1.滿足現有醫療需求可延長1年。
2.進行比較性臨床試驗可延長0.5年。
3.在歐盟境內透過公私協力進行研發可延長0.5年。
若公司另取得比現有療法更有顯著臨床優勢之新適應症,則原有2年市場獨占期可再延長1年,合併計算後保護期共為9.5~11.5年。
而歐盟理事會則不同於歐盟執委會、歐洲議會,選擇將資料專屬保護期維持現行8年,但無法再延長。然而,市場獨占期則提議縮減為1年,但若符合特定條件則可再延長至2年,合併計算後保護期共為9~10年。條件包括:
1.滿足現有醫療需求。
2.產品含有新活性物質,並同時符合以下3項要件:
(1)上市許可申請具關聯性且有證據基礎的比較性臨床試驗結果作為支持。
(2)在一個以上的歐盟成員國內進行臨床試驗。
(3)申請人須證明已優先於歐盟申請上市許可,或於首次向歐盟以外地區提出申請後90日內完成歐盟上市申請。
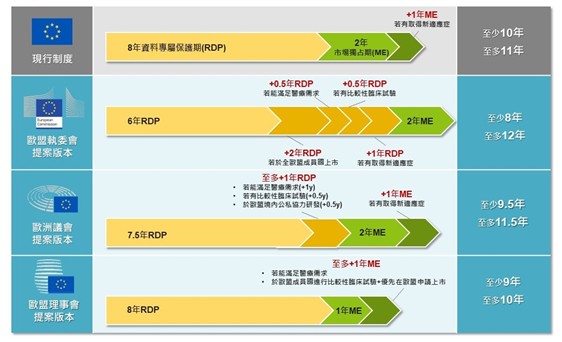
圖一、歐盟藥品法規包裹法案提議版本比較
資料來源:本研究整理
二、針對抗菌藥物引入創新研發機制
抗菌藥物抗藥性(Antimicrobial resistance, AMR)為近年全球公共衛生重要議題,並為世界衛生組織(World Health Organization, WHO)所列全球十大健康威脅之一[6]。因此,除嚴格限制抗菌藥物濫用外,為有效促進新型抗菌藥物之研發,本次藥品法規包裹法案中亦引入「可轉讓資料專屬保護權憑證」(Transferable Exclusivity Voucher, TEV)作為獎勵措施。
使用此憑證可賦予持有人額外1年資料專屬保護期,且除可用於抗菌藥品外,亦可適用於公司內其他更具商業價值藥品,或將此憑證轉賣給其他藥廠,為藥廠提供額外收入,但每張憑證僅能轉讓一次。
考量到憑證可能被用於商業利益龐大之藥品,藉此延緩學名藥與生物相似藥進入市場,並影響病患取得藥物之可及性,歐盟執委會於提議時亦強調此制度預計僅實施15年,或至多核發10張憑證後即停止適用[7]。此外,在歐盟執委會所提議之版本中,對於憑證使用亦設有限制,包含必須於憑證核發後5年內使用,且僅限用於尚在資料專屬保護期前4年之藥品,以確保市場可預測性。
歐洲議會與歐盟理事會雖然大致表態支持,但對憑證效力採取不同立場。歐洲議會態度較為保留,其提議使用憑證所額外延長之資料專屬保護期應依據抗菌藥品優先等級區分為6個月、9個月或12個月,且同樣受有總延長至多1年之限制。換言之,在歐洲議會提議版本下,無論延長事由為何,資料專屬保護期至多僅能延長至8.5年。
相對而言,歐盟理事會並未採納歐洲議會之提議,而是維持歐盟執委會提出增加1年資料專屬保護期之優惠設計。然而,為避免過度限制市場競爭,歐盟理事會亦新增規定,若憑證用於非抗菌藥品,則僅能在該藥品資料專屬保護期的第5年使用,且須證明過去4年內該藥品任一年銷售額均未超過4.9億歐元。
參、總結
「藥品法規包裹法案」反映出歐盟執委會、歐洲議會與歐盟理事會對新藥保護政策之差異,雖然三者皆試圖在鼓勵創新研發與提升藥品可近性之間取得平衡,但在藥品監管保護期間具體設計上仍有差異。歐盟執委會雖提議縮減資料專屬保護期,但也透過延長制度維持研發誘因。而歐洲議會則提議更高的初始保護門檻,但限制總延長期,且更著重在鼓勵研發活動而非市場擴張。至於歐盟理事會則維持現行保護年限,以持續激勵研發投入,但也透過縮短市場獨占期,加速學名藥與生物相似藥上市,以降低藥品價格並滿足醫療需求。
至於「可轉讓資料專屬保護權憑證」制度固然能為抗菌藥物提供研發誘因,但也伴隨著一定的潛在風險與利益分配爭議,如何確保該制度所帶來之公共利益超過公共衛生成本與風險,仍取決於未來最終立法版本應該如何調整與落實。對於我國製藥產業而言,此次改革有可能加速學名藥與生物相似藥進入歐洲市場,亦可能因延長誘因制度而遲延競爭時機,故仍應持續密切關注並審慎調整研發與市場策略。
[1]EUROPEAN COUNCIL, ‘Pharma package’: Council agrees its position on new rules for a fairer and more competitive EU pharmaceutical sector (2025), https://www.consilium.europa.eu/en/press/press-releases/2025/06/04/pharma-package-council-agrees-its-position-on-new-rules-for-a-fairer-and-more-competitive-eu-pharmaceutical-sector/#:~:text=The%20Council's%20position&text=obligation%20to%20supply%3A%20a%20new,patients%20in%20the%20member%20state (last visited Aug. 28, 2025).
[2]EUROPEAN COMMISSION [EC], Reform of the EU pharmaceutical legislation (2023), https://health.ec.europa.eu/medicinal-products/legal-framework-governing-medicinal-products-human-use-eu/reform-eu-pharmaceutical-legislation_en (last visited Aug. 28, 2025).
[3]EUROPEAN PARLIAMENT, Parliament adopts its position on EU pharmaceutical reform (2024), https://www.europarl.europa.eu/news/en/press-room/20240408IPR20308/parliament-adopts-its-position-on-eu-pharmaceutical-reform (last visited Aug. 28, 2025).
[4]「資料專屬保護期」是指專利藥提出新藥上市申請時,所檢附證明藥品安全性與療效之相關申請數據資料,其他藥廠非經原廠同意,於法定保護期間內不得引用,為行政機關針對原廠新藥上市申請資料給予之保護權益。詳請參考:台灣光鹽生物學苑,〈【醫藥小專欄】什麼是資料專屬權(data exclusivity)〉,台灣光鹽生物學苑,https://www.biotech-edu.com/%E3%80%90%E9%86%AB%E8%97%A5%E5%B0%8F%E5%B0%88%E6%AC%84%E3%80%91%E4%BB%80%E9%BA%BC%E6%98%AF%E8%B3%87%EF%A6%BE%E5%B0%88%E5%B1%AC%E6%AC%8A-data-exclusivity/ (最後瀏覽日:2025/08/28)。
[5]「市場獨占期」則是指在一定期間內,即使學名藥或生物相似藥已可向主管機關提出上市許可之申請並獲得核准,但仍不得實質上市銷售,須直到市場獨占期屆滿為止。市場獨占期與資料專屬保護期之比較,詳請參考:Specialist Pharmacy Service, Understanding data exclusivity and market protection (2025), https://www.sps.nhs.uk/articles/understanding-data-exclusivity-and-market-protection/ (last visited Aug. 28, 2025).
[6]〈世界抗生素週〉,衛生福利部疾病管制署,https://www.cdc.gov.tw/En/Category/MPage/t2VWXn93ooNVJPuQRF7jzg (最後瀏覽日:2025/08/28)
[7]CROWELL & MORING LLP, The New EU “Pharma Package”: The transferable exclusivity voucher—A comparison of Commission/Parliament/Council positions(2025),https://www.crowell.com/en/insights/client-alerts/the-new-eu-pharma-package-the-transferable-exclusivity-vouchera-comparison-of-commissionparliamentcouncil-positions (last visited Aug. 28, 2025).




日本內閣官房日本經濟再生總合事務局(内閣官房日本経済再生総合事務局)在2017年6月9日第10次「未來投資會議」中提出未來投資戰略2017報告(未来投資戦略2017~Society 5.0 の実現に向けた改革~),在成長的戰略成果(5)日本第四次產業革命及新經濟的展開中,分別對於機器人實用、物聯網(IOT)、大數據(BIG DATA)、人工智慧(AI)等提出成果及未來計畫。 機器人加速實用化:首先,機器人廣泛利用在商業設施、機場等日常生活空間,於2016年9月羽田機場設置機器人實驗室「Haneda Robotics Lab」,利用機器人改善服務並補充勞動力。有關打掃清潔、協助移動、查詢服務等17種機器人,將進行實證實驗。而路面協助行走型機器人「RT.1」已經完成,於2015年生活協助型機器人之安全性得到國際認證,其後發展之「RT.2」將使用於長期照顧層面。其次,開發農業使用之自動駕駛拖車,並提供工作實際狀況和土壤狀況之電子管理服務。今年6月開始商業化之自動駕駛顯示器,可以監控自動駕駛耕作機器進行自動耕作等。在物流管理方面,於2018年將於山間部等地區進行無人機的包裹遞送,2020年將在都會區全面無人包裹遞送。預計將與日立等相關公司,進行物流管理系統之開發及活用福島機器人測試場域。
日本公告修正電業法(電気事業法),將放寬電力工作物定義,並新增電力事業類型日本於2020年(令和2年)6月12日公告修正「電業法」(電気事業法,以下簡稱新電業法),並預計於2022年(令和4年)4月1日正式施行。本次修正有以下兩個重點:(1)調整電力工作物之定義;(2)新增電力事業之種類,以符合國際技術及市場之變遷。 日本新電業法調整了電力工作物之定義,將儲能設備納入了電力工作物之定義中,日本新電業法下所謂電力工作物係指:「為了發電、儲能、變電、輸電、配電或其他以電力使用為目的而設置之機械、器具、水壩、水道、蓄水池、電路、電線或其他工作物(但船舶、車輛或航空器等,則依其他法令定之)。」此外,日本新電業法也於電力事業之種類下,新增了「特定電力批發業」(特定卸供給業)此一新型電力事業。依日本新電業法對於「特定電力批發業」之定義,所謂「特定電力批發業」,係指:「自其他發電及儲能設備設置者(但不包含發電業者)處聚合其發出或放出之電力而再向售電業、一般輸配電業、輸電業、特定輸電業供應電力者。」又依日本經濟產業省規劃,「特定電力批發業」得自行設置儲能設備,自產消合一之用電戶(prosumer)之儲能設備或發電設備(包含但不限於再生能源)購入電力,並轉而再至電力交易市場向其他售電業者販售電力;但「特定電力批發業」不得直接從發電業者或其他售電業購買電力,也不得直接售電予用電戶,故特定電力批發制度實際上是旨在賦予聚合商(アグリゲーター)參與市場之地位。
英國Patent Box租稅優惠政策英國Patent Box租稅優惠政策 科技法律研究所 2014年03月25日 壹、政策背景 歐盟在2000年規劃的里斯本策略[1],就創造工作機會與保障勞工權益方面積極作為,驅使歐盟各國稅法改革,成功使歐盟在國際上擁有強大稅制上競爭力。而後法國、匈牙利、盧森堡、比利時、荷蘭、西班牙等國家,先後就研發活動施行優惠稅制,更強化其研發活動創造、發展的誘因。 2011年英國政府成長計畫[2]檢討近10年來,各國降低稅率、打破貿易壁壘、培育專家人才,同時英國經濟疲弱,負債且競爭力下降。因此英國政府在計畫中提出要建立G20[3]中最具競爭力之稅制。在不願與其他國家進行稅金削價競爭下,英國政府決定自專利著手,認為專利與高科技研究發展最具關連性,且專利本身已具完備審查機制,於是以專利獲利為主的優惠稅制逐步成形,稱為Patent Box,2011年6月英國財政局HMRC廣納各界意見,並在2012年Finance Act修法草案第8A章節提出。 對於Patent Box的設計,英國政府提出幾個原則:首先為不造成企業困擾,Patent Box必須為自願性參加;並且2013年4月起,為來自專利的獲利提供10%的優惠稅率;專利獲利之計算應避免為企業製造不必要的行政成本;不以總收入計算而以淨獲利計算;鼓勵持續研發等。 貳、政策方針 一、政策目的 英國政府早期對法人的租稅獎勵,係針對中小法人購置網際網路軟硬體設備、研發相關費用等,提供100%當年度投資抵免的獎勵;而為促進法人進行科學技術研究,2000年開始提供中小法人投入研發投資抵減獎勵,2002年擴及於大法人亦得適用,但抵減率降為25%。 英國政府為了鼓勵法人將高值專利與其運用發展紮根國內,以法人稅(Corporation Tax)角度出發,提議租稅優惠方案「Patent Box」。政府認為此方案能夠提供國內法人額外動機,將高價值工作與專利相關生產活動留在國內,並強化英國法人目前在全球的高值研發能量。 此方案由工黨政府提出,但直到2010年11月保守黨政府執政時,才將之納入執政提議政策,隔年6月與幕僚單位諮詢相關立法細節與範圍後,迅速於同年12月擬出草案。2012年7月,Patent Box方案經皇家同意被納入「法人稅法(the 2010 Corporation Taxes Act)」之修法法案「財政法(the 2012 Finance Act)」之中。2013年4月起,英國法人若透過自主研發或委託研發所產生之專利而賺取之收入,法人稅可由原本的20~24%(2013年4月法人稅為23%),調降適用10%的優惠稅率。 此方案的推出,一部份原因為產業界輿論稱英國原租稅制度使法人逐漸喪失競爭力;而之所以將租稅優惠鎖定為專利權(與特定植物品種權),係因為專利權與高科技研發生產具有很強的鍊結,並且專利權本身的審核制度更已為Patent Box方案挑選出真正的創新科技發明。 二、Patent Box 優惠稅制影響評估 (一)英國國庫 英國Patent Box採逐步調降稅率之實施方式,以減輕對國庫的負擔,2013年以獲利的60%適用Patent Box 10%的優惠法人稅稅率,之後隔年增加10%,直至2017年4月1日,100%獲利適用10%之優惠法人稅稅率。而HMRC預算預估之英國專利獲利金額,係以其資料庫為基礎推算之。 (二)英國法人 調降法人稅對英國法人而言,也降低了將相關專利獲利移出境外之動機;此外,對於未申請專利者,Patent Box更增加其申請的動機,繼而提高發明之價值。 對於中小型法人而言,此方案可使其檢視自身專利布局,但對於未接觸過專利想加入此方案者,會增加該些中小型法人之相關行政成本,但Patent Box一產品僅須具一專利之設計,可減輕對此類法人之負擔調降法人稅對英國法人而言,也降低了將相關專利獲利移出境外之動機;此外,對於未申請專利者,Patent Box更增加其申請的動機,繼而提高發明之價值。 對於中小型法人而言,此方案可使其檢視自身專利布局,但對於未接觸過專利想加入此方案者,會增加該些中小型法人之相關行政成本,但Patent Box一產品僅須具一專利之設計,可減輕對此類法人之負擔。 (三)英國經濟 由專利獲利的法人可受惠於此方案,尤其是醫藥、生技、電子、國防等產業。Patent Box方案更可吸引國外創投資金,增進國內經濟並增加高值工作機會。 [1]Directorate-General for International Policies - Policy Department, The Lisbon Strategy 2000-2010, European Parliament (2011), http://www.europarl.europa.eu/document/activities/cont/201107/20110718ATT24270/20110718ATT24270EN.pdf (last visited, Feb 19, 2014) [2]HM Treasury, The Plan for Growth, March 2011, https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/31584/2011budget_growth.pdf (last visited, Feb 11, 2014) [3]G20,一個國際經濟合作論壇,由八國集團(美國、日本、德國、法國、英國、義大利、加拿大和俄羅斯)以及其餘十二個重要經濟體(歐盟、中華人民共和國、巴西、印度、澳大利亞、墨西哥、韓國、土耳其、印尼、沙烏地阿拉伯、阿根廷和南非)組成。
歐盟執委會就中國大陸法院頻發禁訴令情形,循WTO爭端解決機制向中國大陸提出諮商請求歐盟執委會(European Commission, EC)於2022年2月18日向中國大陸提起諮商請求(request for consultation),此係世界貿易組織(World Trade Organization, WTO)爭端解決機制(Dispute Settlement Mechanism, DSM)之一環,並依照「爭端解決程序與規則瞭解書」(Understanding on Rules and Procedures Governing the Settlement of Disputes, DSU)第4.4條之規定,副知WTO的爭端解決機構(Dispute Settlement Body, DSB)。 EU於此諮商請求中指出,自2020年8月中國大陸最高人民法院就Huawei對 Conversant案此一涉及標準必要專利(Standards-Essential Patents, SEP)之訴訟核發禁訴令(Anti-Suit Injunction, ASI),並裁定違反者將被處以每日100萬人民幣之罰款後,中國大陸各法院即頻以此方式禁止外國專利權人於中國大陸以外之法院提起或續行專利訴訟(例如:Xiaomi對Interdigital案、ZTE對Conversant案、OPPO對Sharp案、Samsung對Ericsson案等)。中國大陸法院此等措施(measures)限制歐盟會員國公司行使專利權,違反「與貿易有關的智慧財產權協定」(The Agreement on Trade-Related Aspects of Intellectual Property Rights, TRIPS);關於此,EU先前已多次與中國大陸政府交涉,例如曾在2021年7月依據TRIPS第63.3條(會員國法律文件透明化要求),發函請求中國大陸公開專利相關判決之進一步資訊,然均未果。故EU此次提出與中國大陸政府諮商之請求。 EU此諮商請求是DSM的第一步;如雙方未能於中國大陸政府收到諮商請求之60日内解決爭端,依照DSU第4.7條,EU得要求DSB組成一專門小組,就此事進行審理,以認定中國大陸多數法院頻發ASI一事是否違反TRIPS第1.1條、第28.1條、第28.2條及第41.1條、第44.1條及第63.1條等。