新德國包裝法簡介

為有效降低包裝廢棄物對環境造成的汙染及不利影響,使製造商履行其B2C(business to customer)產品責任,德國以新的包裝法(Packaging Act, VerpackG)取代現行的規範(Packaging Ordinance,VerpackV),並已於2019年1月1日生效。
新包裝法VerpackG不同於VerpackV之處,在於除要求業者須加入原有的回收系統外,另授權Zentrale Stelle(Stiftung Zentrale Stelle Verpackungsregister,ZSVR)基金會作為新包裝法強制登記制度的執行單位,規範欲在德國銷售產品包裝之所有實體或網路製造商及零售商,有義務於ZSVR的數據資料庫”LUCID”註冊,才能在德國地區進行銷售,並且為全面提升透明度,乃規範於LUCID註冊之商家資訊皆屬可供大眾公開查詢。
依VerpackG規定,於2019年1月1日起未為註冊的商家,其包裝商品不能在德國上市,否則恐將臨100,000歐元之罰款;另未加入回收系統之商家,恐面臨200,000歐元之罰款。而除須註冊與回收系統的加入外,製造商及零售商尚須將以下之包裝相關資訊提供給ZSVR做比對:
(一)註冊號碼(商家於資料庫註冊時,由ZSVR所提供之註冊號碼)
(二)包裝材料及容積
(三)製造商履行生產者延伸責任(Extended Producer Responsibility)簽訂的包裝方案名稱
(四)與回收公司或回收系統間簽訂之契約期限
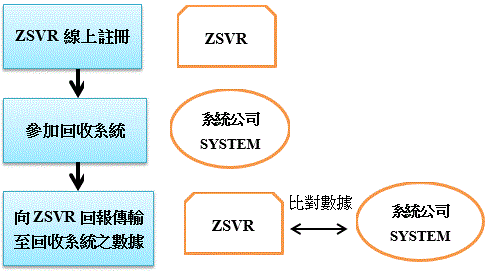
資料來源:自行繪製
圖 德國包裝法實施步驟

蔡宛君
法律研究員 編譯整理
How-To Guide to the Packaging Act for Manufacturers, Stiftung Zentrale Stelle Verpackungsregister(2018), https://www.verpackungsregister.org/fileadmin/user_upload/How-to-Guide_en_13072018_final.pdf (last visited Jun. 1, 2019)
The ten most important questions regarding the implementation of the Packaging Act ,Stiftung Zentrale Stelle Verpackungsregister(2018), https://www.verpackungsregister.org/fileadmin/user_upload/10-W-Fragen_en_13072018_final.pdf (last visited Jun. 1, 2019)
The new German Packaging Act is here – and it’s particularly important for online retailers,Der Grüne Punkt, https://www.gruener-punkt.de/en/services/packaging/german-packaging-act.html (last visited Jun. 4, 2019)



美國聯邦貿易委員會(FTC)針對國家公路交通安全管理局(NHTSA)的行政命令提出建議,就有關車輛到車輛通信(V2V)之事宜,FTC長期作為負責保護消費者隱私與安全的聯邦機構,FTC認為NHTSA在行政命令中採取隱私和安全問題考慮是非常適當。 在FTC的建議評論指出,FTC針對物聯網的的資訊安全疑慮,同樣也會適用在消費者的車輛收集的隱私和安全問題。FTC認為NHTSA的協商支持作法,基於流程的可解決隱私和安全隱患,其中包括隱私風險評估。該評論還讚揚NHTSA設計一個V2V系統來限制收集和存儲僅是供應其預期的安全目標的數據。 美國每年都會有上千人意外死於汽車意外事故,NHTSA研究指出,汽車相撞的原因多數情況下在於資訊的不透明,如果汽車之間可以「相互溝通」,讓駕駛彼此知悉對方的情況,就能減少碰撞事故。 「V2V」係指vehicle-to-vehicle,是規劃建立於汽車之間的通信網路。在這個網路中,汽車之間能夠互相傳送數據,告訴對方自己的狀態和行為,也了解其他車輛的狀態和行為。但是目前V2V各家發展的標準不一,因此假設福特的車如果不能跟其他廠商的汽車溝通,技術再好也沒用。 也因此,NHTSA在官網上公告規則,宣布將制定「V2V」通信技術標準的法規。也就是說,NHTSA將要制定一個統一的標準,來確保汽車之間溝通使用的是同一種語言。在最新的一份報告中,NHTSA詳細說明了「V2V」通信技術的軟硬體標準。它包括部署該項技術可能需要的硬體設施及其費用,汽車之間溝通的資料類型,以及該技術將如何提醒司機。此外,還覆蓋了「V2V」通信技術的安全細則,以及它將如何加密以避免竊聽和侵犯隱私。 在使用者和廠商都關心的資料外洩方面,NHTSA表示,資料本身將不包含個人身份資訊,並且將會被保密。目前提出的方案裡包含兩套數據,其中一個包含核心資訊:如位置、速度、駕駛方向、剎車狀態、車輛尺寸等。這些資料將即時更新並相互傳播。第二套數據則會更加複雜,只有在數據發生變化時才會相互傳輸。它包括汽車輪胎是否漏氣,前燈是否打開,保險杠的高度,是否行駛在密集人群中等。
歐盟執委會發布「歐洲健康資料空間」規則提案,旨在克服健康資料利用之障礙歐盟執委會(European Commission)於2022年5月3日發布「歐洲健康資料空間」(European Health Data Space, EHDS)規則提案,其旨在克服健康資料利用之障礙,以充分發揮數位健康與健康資料之潛力。EHDS為一個專門用於健康之資料共享框架(health-specific data sharing framework),針對患者以及用於研究、創新、政策制定、患者安全、統計或監管目的等電子健康資料之運用,建立明確規則、通用標準與實務、基礎設施與治理框架,無論是個人、醫療人員、健康照護提供者、研究人員、監管人員、產業界皆可由此受益。 EHDS之具體內容主要包括九個章節: (1)第一章為一般條款(General provisions),內容包括本規則之主題與範圍,並闡明定義、以及與其他歐盟法規之關係; (2)第二章為電子健康資料之原始利用(Primary use of electronic health data),其針對歐盟一般資料保護規則(GDPR)所載權利,增訂補充性之配套保護機制,並設定醫事人員及其他健康從業人員針對EHD之義務; (3)第三章為EHR系統與福祉應用(EHR systems and wellness applications),其主要重點為EHR系統之強制性自我認證計畫(mandatory self-certification scheme),要求其需符合可互通性與安全性等基本要求,並界定EHR系統中各經濟營運商(economic operator)之義務、EHR系統合規(conformity)要求,並負責EHR系統市場監督機構之義務; (4)第四章為電子健康資料之二次利用(Secondary use of electronic health data),如將資料用於研究、創新、政策制定、患者安全或監管活動。本章定義一組資料類型,規範可利用之既定目的以及受禁止之目的(如商業廣告、增加保險、開發危險產品),並規定會員國必須建立健康資料近用機構(health data access body),以便電子健康資料的二次利用,並確保由資料持有者所產生之電子資料可提供給資料使用者; (5)第五章為其他行動(Additional actions),其旨在提出其他措施以促進會員國之能量建構(capacity building),以配合EHDS之發展,包括數位公共服務之資訊交換、資金,並規範於EHDS下非個人資料之國際近用規定; (6)第六章為歐洲治理與協調(European governance and coordination),其創建「歐洲健康資料空間委員會」(European Health Data Space Board, EHDS Board),促進數位健康當局及健康資料近用機構之間的合作,特別是電子健康資料之原始與二次利用間之關係,並包含歐盟基礎設施聯合管理小組(joint controllership groups for EU infrastructure)相關規定,其任務在於就電子健康資料之原始與二次利用所需之跨境數位基礎建設進行相關決策; (7)第七章為授權與委員會(Delegation and Committee),其允許歐盟執委會通過關於EHDS之授權法案(delegated acts),並希望根據C (2016) 3301號決定成立一個專家小組,以便於準備授權法案、實施本規則時提供建議與協助; (8)第八章為附則(Miscellaneous)規定,其中包含關於合作與處罰之規定,以及要求於本規則實施後進行評估與檢視之條款; (9)第九章為延遲適用與最終條款(Deferred application and final provisions),其規定本規則與個別條款之生效日。
台美貿易談判 藥廠權益是焦點據報載, 5 月 25 日 起在我國舉行兩天之台灣與美國貿易投資架構協定( TIFA )會談,藥廠權益乃雙方談判焦點,美方這次來台所提出之談判項目中,對台灣藥廠衝擊較大的是資料專屬權( Data Exclusivity ),及專利連結( Pattern Linkage )兩項,本土製藥業擔心,政府若妥協將可能造成台灣藥廠及研究單位新台幣上百億元的損失。 儘管去年初立法院已經三讀通過藥事法 40 條之 2 的「資料專屬權保護」條文,但預料美方這次將要求政府重新修法,以保障外商藥廠的權益。此外,專利連結( patent linkage )也是衛生署嚴陣以待的項目,外商訴求此一機制之目的,係希望透過專利資訊之揭露,使任何申請上市許可之學名藥品,均係在專利到期後或未侵害專利之前提下,使得上市。 專利連結制度首見於美國,美國食品藥物管理局 (FDA) 對藥品有所謂之「橘皮書」,要求公布各藥品的專利內容及安全性與療效資訊,並以此作為日後學名藥賞上市或與原開發藥廠發生專利侵權爭訟時之參考。業界認為,如果台灣也比照美國 FDA 專利連結的規定,可能導致外商藥廠得以輕易對台灣藥廠展開侵權訴訟官司,衝擊我國製藥產業。
IBM嘗試新方法支持開放原始碼IBM公司在2日拉斯維加斯舉行世界夥伴(PartnerWorld)會議時,宣布提倡開放原始碼創新的新措施,包括成立求職應徵者資料庫,以及一項電子學習計畫。這座資料庫預定今年第三季推出,屆時會把具有開放原始碼技術的大學生所投的履歷表一一編列成目錄。想被納入資料庫的資格,包括曾經參加IBM校園人才培訓計畫(Academic Initiative)中級程度以上,並通過IBM開放原始碼專業資格考試的人士。該資料庫提供IBM的企業客戶與商業夥伴檢索。起初,此資料庫只涵蓋北美洲地區,但IBM打算將來擴大推廣到世界其他地區。 該公司也將透過提供IBM校園人才培訓計畫,提供各校所需的中介軟體及硬體,而Hubs計畫本身不打算收費,或只酌收少許費用。第一座這種中心預定春季在德州A&M大學成立。 IBM另外在PartnerWorld宣布,計劃今年與商業夥伴共同成立100座新的「創新中心」( innovation centers)。藍色巨人先前已承諾投資1.5億美元開辦這類中心,讓系統整合業者、獨立軟體公司、附加價值流通業者以及解決方案服務提供者藉此取得IBM的技術與設備,以協助他們測試並最佳化自家產品。其構想是協助這些夥伴加速產品上市,並降低產品開發費用。自2004年推出以來,IBM已在北美和歐洲成立大約40座這種中心。