德國第二部開放資料法案簡介

德國第二部開放資料法案簡介
資訊工業策進會科技法律研究所
2023年03月25日
壹、前言
德國聯邦政府於2021年7月16日通過「第二部開放資料法案」(Zweiten Open-Data-Gesetz),包含修正《電子政務促進法(電子化政府法)》(E-Government-Gesetz,EGovG)第12a條,以及修正《資訊再利用法》(Informationsweiterverwendungsgesetz,IWG)並更名為《公部門資料使用法》(Datennutzungsgesetz - DNG),並於同年月23日生效。
修正目的係為充分發揮資料開放政策之潛力,建立一個初步的監管框架,改善資料提供、提高標準化及互通性的問題,以進一步提高資料之可用性。另外,也為轉換歐盟於2019年6月修正《開放資料與公部門資訊再利用指令》(DIRECTIVE (EU) 2019/1024,Open Data and the re-use of Public Sector Information Directive,下稱開放資料指令)[1]。
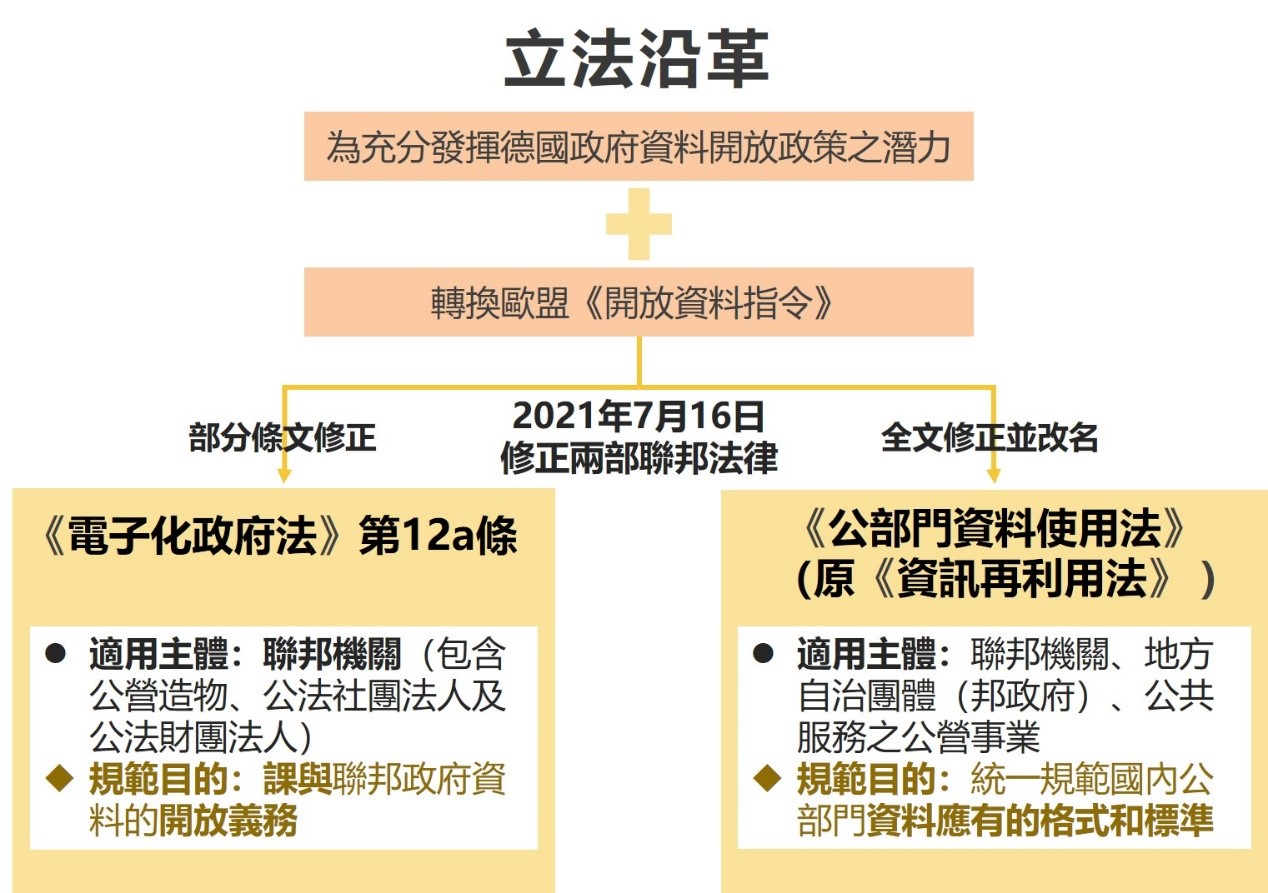
資料來源:作者自繪
圖一 「德國第二部開放資料法案」立法沿革
以下分別介紹電子化政府法第12a條及《公部門資料使用法》的修法內容:
貳、修法內容
一、電子化政府法第12a條
2017年7月13日,德國在《電子化政府法》新增第12a條「聯邦直接行政機關開放資料」(Offene Daten der Behörden der unmittelbaren Bundesverwaltung)規定,做為「聯邦政府」進行開放政府資料的法源[2]。本次修法法案,調整電子化政府法第12a條的部分條文,並改稱標題為「聯邦開放資料授權命令」,課予聯邦政府之政府資料開放義務(但仍未賦權民眾依據本法要求開放特定之政府資料)。以下簡介新舊規範比較:
(一)受規範主體範圍:所有聯邦機關
舊法限於「直接從屬於聯邦政府的行政機關」,不包含自治團體(如邦政府)、公立機構、公立法人、公立基金會、公立大學等[3]。
新法則擴大主體範圍,不限於「直接從屬於聯邦政府的行政機關」,而涵蓋所有「聯邦機關」(Die Behörden des Bundes)(第1條),包含公營造物、公法社團法人及公法財團法人等[4];這些新適用主體開放資料的緩衝期,為法規生效後12個月內應首次提供。不過,適用主體仍不包含自治團體(如邦政府)與公權力受託人[5]。
(二)受規範客體範圍
同舊法,本法所稱的資料,仍為「由聯邦機關為完成公共任務所蒐集,或由受委託之第三方代表聯邦機關所蒐集之資料」,且包含「研究資料」,並應符合以下5款條件:
1.以電子方式儲存、以集合形式結構化(尤其適用於列表和表格)。
2.存在於該機關之外的事實,但與該機關有關者
3.非由聯邦機關處理其他資料所產生的結果
4.調查後未經過處理者
但有以下情形時可以處理後再開放:(1)為糾正錯誤而處理;(2)基於法律或事實原因而處理,例如依個資法規定匿名)。
5.資料涉及個人時,匿名化且無法再識別個資者。
舊法中,以負面表述的方式,提及涉及個資保護事由時,不開放資料;新法則增加正面表述,資料雖涉及個人,但若已去識別化且無法再識別者,即可作為開放客體。
(三)開放原則及例外
與舊法相同,採取「預設開放原則」(Open by Default),即已經完成電子化,且不具例外理由時,聯邦機關即須開放這些資料。
例外則是符合以下情形者,聯邦機關不須提供資料:
1.資訊自由法規定不公開之政府資料
根據資訊自由法(Informationsfreiheitsgesetz,IFG)第3、4及6條(涉及特殊公共利益的資料、保護官方決策程序的資料、保護智慧財產權及營業秘密),不具取用權(Zugangsrecht,right of access)或僅具有限取用權的資料(與舊法相同)。
2.只有在第三方參與後,取用權才存在的資料(與舊法相同)
例如依據相關法規規定,須經過聽取第三方意見的程序(如聽證會),或須衡量第三人利益,才能提供資料的情形。[6]
3.資料由非受公權力委託之第三方建置,且非法律規定有傳輸義務者(與舊法相同)
4.已透過其他管道開放之研究資料(新法新增)
基於研究目的所蒐集之資料,且已透過開放取用(open access)的網路免費提供者,無須再提供;但仍可自願於國家詮釋資料入口網站(GovData)開放。
此為新法新增,不同於舊法,「基於研究目的所蒐集之資料」應開放提供,但法規特別規定如果已經基於其他原因已可開放取用者,即依照原先狀態繼續開放取用,機關無義務再透過其他管道提供[7]。
5.受銀行保密義務保護之資料(新法新增)
6.含有個人資料的資料集
特別從前款「資訊自由法規定不公開之政府資料」拉出,單獨成項(新增於電子化政府法第12a條第3a項)作為強調。
(四)資料提供方式
1.原則蒐集後立即提供,但研究資料可延後
新法新增為研究目的所蒐集之資料的提供時點彈性,研究資料若要提供,則應於研究計畫完成且研究目的完成後才能提供[8]。
2.以機器可讀格式提供
3.提供詮釋資料於「國家詮釋資料入口網站」(GovData)
4.免費、隨時、無須理由、不須註冊、透過公開使用網路提供資料檢索
5.可不受限制的再利用
(五)各部門設立協調員與中央辦公室聯繫
聯邦政府應設立中央辦公室,負責就聯邦機關提供資料作為開放資料時給予建議,並作為各邦負責開放資料辦公室的聯絡點。
聯邦各機關也應任命一名開放資料協調員,作為各部門與中央辦公室聯繫的聯絡人,並致力於識別、提供及再利用其所屬部門之開放資料。
(六)推行開放資料
與舊法相同,聯邦政府應每2年提出研究報告,向聯邦眾議院報告透過聯邦機關(不包含公營事業)開放資料的進展。
新法另增訂兩項事宜:
1.聯邦政府應評估開放資料法之適用主體擴展到自治團體、邦政府、受委託行使公權力的自然人與法人的可能性。
2.授權聯邦內政部與其他聯邦權責單位及聯邦政府專員達成協議,訂定法規命令規範開放資料之具體作法。
(七)公務員免責
聯邦機關沒有義務檢查所提供的資料的正確性、完整性、可信性。
(八)第12a條不能作為提供資料的請求權依據
電子化政府法於第12a條第1項後段明文該條不能作為提供資料的請求權依據,因此人民未被賦予得以請求政府開放資料之權利。[9]
二、公部門資料使用法
德國於2006年轉換2003年版的歐盟《公部門資訊再利用指令》(Directive 2003/98/EC,下稱PSI指令),制定《資訊再利用法》(Informationsweiterverwendungsgesetz,IWG),並隨著PSI指令於2013年的修正(Directive 2013/37/EU)而於2015年修正。本次再基於PSI指令於2019年修正為開放資料指令,《資訊再利用法》隨之全部修正,並更名為《公部門資料使用法》(Datennutzungsgesetz - DNG),以下簡介規範內容:
(一)立法目的
《公部門資料使用法》的立法目的為「政府資訊再利用」、「改善公部門資料在商業目的之使用」,而統一政府資料的格式、授權、開放原則、收費原則及非歧視原則等內容。公部門資料使用法第1條第2項明文強調,本法未課予提供資料之義務或賦予近用資料之權利。
另外,本法係用以統一國內各相關法規之規範標準,使聯邦及各邦的資料,都能順利被用於商業及非商業利用[10]。
(二)適用主體
1.公部門機構
包含聯邦機關、地方自治團體(如邦政府),及公營造物、公法社團法人及公法財團法人等。
2.提供公共服務之公營事業
本次修法,納入提供公共服務(例如能源及交通)之公營事業,這些公共服務公營事業包含獨佔事業及處於競爭市場者。
3.提供研究資料的大學、研究機構及研究資助機構
(三)適用客體
1.一般政府資料
公部門機構與公共服務依法有義務提供之資料、依法有近用權之人可近用之資料。
2.高價值資料集
公部門機構與公共服務,若要提供歐盟開放資料指令之6種主題類別高價值資料集時,須透過適當之API、機器可讀、可批量下載的方式提供。
3.研究資料
研究資料係指,由公共資助的科學研究活動中所蒐集,或作為證據使用,或用以驗證研究成果的數位形式紀錄。且已透過大學、研究機構、研究資助機構、所屬研究人員或計畫資料庫開放取用(open access)。
若研究資料與合法商業利益、知識轉移活動或第三方智慧財產權等原因相衝突,則不適用本法方式提供。
4.不適用客體
與個資保護相衝突的資料、與商業秘密相衝突的資料、國家安全及公共安全資料、關鍵基礎設施保密資訊、保密統計資料、涉及第三人智慧財產權資料、不屬公部門機關公務範圍之資料、標誌及徽章、公共節目及廣播、文化機構資料、中學以下教育機構資料、中學以上教育機構非研究資料。
(四)提供格式
1.提供既有之可利用格式資料
聯邦機關或公營事業,只須提供既有的、已電子化、機器可讀、FAIR資料原則(可近用、可搜尋、可再利用、可互通格式)、符合開放標準(確保可互通)的資料。
2.詮釋資料
機器可讀的詮釋資料,應透過國家詮釋資料入口網站(GovData)提供。
3.動態資料
如果資料具有波動性或時效性的特性,而須頻繁或即時更新時,則聯邦機關或公營事業應透過API及批量下載方式,在資料蒐集後立即提供。
(五)授權提供
原則上不一定要以授權方式提供資料,但一般公立圖書館、大學圖書館、博物館及檔案館中具有智慧財產權的資料,以及提供公共服務的公營事業的資料,皆應以授權方式提供。使用授權條款時,應盡可能使用開放授權,或不做非必要的使用限制。
(六)資料原則
1.「概念上與標準化之開放」原則
應盡可能依據「概念上與標準化之開放」原則(der Grundsatz „konzeptionell und standardmäßig offen“)建置本法規範範圍的資料。立法理由稱,依此原則,本法適用之資料應盡可能以「預設開放」方式提供[11]。
2.收費原則
一般聯邦機關,應免費提供資料,但允許因以下原因收取邊際成本補償費用,且收費標準應透明公開:a.資料複製、提供及傳輸;b.個資匿名化措施;c.營業秘密保護措施。
例外可收取費用的機關則有以下3類,且收費標準亦應透明公開:a.須自籌財源履行公務委託之機關;b.一般公立圖書館、大學圖書館、博物館及檔案館;c.提供公共服務的公營事業。
所有聯邦機關及提供公共服務的公營事業,提供高價值資料與研究資料時,應免費且不得收取邊際成本補償費。但一般公立圖書館、大學圖書館、博物館及檔案館提供高價值資料與研究資料時,可收取費用。
3.非歧視原則及禁止排他原則(不得與他人訂定專屬授權)
參、法律適用關係
(一)電子化政府法之法律適用通則規定
電子化政府法第12a條與其他相關法規涉及「資料」規定之間的關係,須依據電子化政府法第1條第4項法律適用通則規定處理,該條項規定「當有相同內容的規定或相衝突的規定存在時,不適用本法」,因此在電子化政府法第12a條與其他相關法規的適用優先順序上,僅在沒有聯邦法律對於「資料提供」有相同或相矛盾的規定情況下,才能適用電子化政府法第12a條,反之,當有任何其他法規涉及「資料提供」時,電子化政府法第12a條就退居於次位,以使其他法規的立法目的仍可實現,也能避免與國際標準或歐盟法規的要求重複、矛盾或衝突[12]。
(二)電子化政府法第12a條與公部門資料使用法之適用關係
雖然公部門資料使用法未課予提供資料之義務或賦予近用資料之權利,但由於仍規範資料之標準及格式,使政府資料能再利用,所以仍被認為涉及「資料提供」,而有法律適用優先順序問題。
因此,電子化政府法第12a條開放資料法的適用主體及客體,基本上適用公部門資料使用法的相關規定,包含政府資料的格式、授權、開放原則、收費原則及非歧視原則等內容。例如:圖書館所藏資料,若涉及高價值資料的提供時,圖書館可收取費用。
肆、評析
德國聯邦政府透過於電子化政府法制定開放資料法相關規範,除了促使聯邦政府機關將政府資料全部改以電子化、機器可讀方式建置以外,也促使政府資料以「預設開放」為原則建置,使政府資料能更被加值活化再利用。
而2021年的修正,更將「基於執行政府出資研究計畫目的所蒐集之資料」,以及「公營造物、公法社團法人及公法財團法人等主體所建置的政府資料」納入開放客體,擴大開放政府資料的範圍。
保護機敏資料部分,除了原先規範得不開放具第三方智慧財產權之資料,以及具國家安全、特殊公共利益等資料以外,新修正法案也加強規定個資保護(含有個人資料之資料集不開放,僅開放已匿名化資料),以及增列「受銀行保密義務保護之資料」為不須開放資料。
另外新修正的法案,也加上許多推行開放資料的措施,包含「聯邦各部門應任命一名開放資料協調員」、「開始評估政府開放資料義務要求,擴展到邦政府及受委託行使公權力的可能性」,以及「授權政府開放資料主管機關聯邦內政部,訂定法規命令規範開放資料之具體作法」等。
綜上,德國透過開放政府資料專法及公部門資料使用法,建立開放資料的格式標準及開放原則,帶頭推動國內的資料開放環境,以促進資料的進一步活化再利用。
[1]Council Directive 2019/1024, art. 17, 2019 O.J. (L 172) 56, 77.
[2]立法理由A、IV、第1段:「聯邦行政對提供資料開放之監管,就其性質而言,屬於聯邦之獨立事務,因此僅能由聯邦政府本身監管。」Deutscher Bundestag, BT-Drucksache 19/27442 (Gesetzentwurf der Bundesregierung), S. 19, https://dserver.bundestag.de/btd/19/274/1927442.pdf (last visited Mar. 21, 2023).
[3]Wiss. Mit. Heiko Richter, „Open Government Data“ für Daten des Bundes - Die Open-Data-Regelung der §§ 12 a, 19 E-Government-Gesetz, NVwZ, 2017(19), 1408, 1409 (2017).
[4]立法理由A、II、第1段。Deutscher Bundestag (Fn. 2), S. 17. https://dserver.bundestag.de/btd/19/274/1927442.pdf (last visited Mar. 21, 2023).
[5]Deutscher Bundestag (Fn. 2), S. 2.
[6]Von Dr. Ralf Schnieders, Die neue Open-(Government)-Data-Gesetzgebung in Frankreich und in Deutschland, Die Öffentliche Verwaltung, 2018(5), 175, 183 (2018); Deutscher Bundestag, Drucksache 18/11614 (Gesetzentwurf der Bundesregierung), S. 20, http://dipbt.bundestag.de/dip21/btd/18/116/1811614.pdf (last visited Mar. 21, 2023)
[7]Deutscher Bundestag (Fn. 2), S. 30.
[8]Deutscher Bundestag (Fn. 2), S. 30-31.
[9]Wiss. Mit. Heiko Richter (Fn.3), S. 1412.
[10]Deutscher Bundestag (Fn. 2), S. 18.
[11]Deutscher Bundestag (Fn. 2), S. 33.
[12]Deutscher Bundestag, Drucksache 18/11614 (Gesetzentwurf der Bundesregierung), S. 17, http://dipbt.bundestag.de/dip21/btd/18/116/1811614.pdf (last visited Mar. 21, 2023); Wiss. Mit. Heiko Richter (Fn.3), S. 1412.





加州聯邦中區地方法院於2014年6月在Jancik v. Redbox Automated Retail, LLC (No. SACV 13-1387-DOC, 2014 WL 1920751 (C.D. Cal. May 14, 2014))一案中,判決影片自動出租機公司Redbox勝訴。法院認為,雖然Redbox在其經營的線上影視串流服務中未提供隱藏字幕(closed captioning),導致聽障者無法藉由閱讀影片字幕來了解劇情,但「網站」非美國身心障礙者法(Americans with Disabilities Act,下稱ADA)第三章「民間事業體所營運之公共設施與服務」中所稱「公共設施」(public accommodation),即無障礙建置範疇不包含提供公眾商品與服務的「網站」,因此業者不須提供具可及性之商品,例如:附字幕影片。法院認為第三章並未就公共設施中商品特色和內容有所規範,因此業者無義務改善其他影片存貨規格,使其能為身障者所觀看;又Redbox線上影視串流服務僅有網路通路,依ADA文義解釋,網站亦非屬於公共設施,無提供無障礙建置之必要。 本案與第一巡迴上訴法院在NAD v. Netflix案見解大相逕庭,該案以「美國國家聽障人士協會」(National Association of the Deaf, NAD)為首之公協會,集體對美國知名線上串流影視節目網站Netflix提起訴訟,控告其線上影視節目未提供隱藏字幕,使得聽障人士無法觀看該影片內容,法院判決該平臺網站屬於「公共設施」,依ADA第302條規範,身心障礙者有權利享受公共設施之設備,不得因殘障而受差別對待。有關網站是否屬於ADA第三章所稱公共設施,而使得私法人有改善網頁無障礙技術義務,仍有待觀察。
新加坡個人資料保護委員會針對企業蒐集、使用、揭露永久居留證(NRIC)號碼提出新的諮詢指引考量各行各業的從業習慣及民眾對企業蒐集、使用、揭露永久居留證(National Registration Identification Card, NRIC)號碼之看法,新加坡個人資料保護委員會(Personal Data Protection Commission, PDPC)於2017年11月提議修改個人資料保護法的諮詢指引(Advisory Guidelines on the Personal Data Protection Act ),明確界定企業蒐集、使用、揭露NRIC及其號碼之範圍。 依據舊的諮詢指引,新加坡個人資料保護法允許企業在基於合理特定目的並依法獲得當事人有效同意之情況下,蒐集、使用或揭露NRIC號碼。因此,不少企業活動習慣蒐集利用民眾的NRIC號碼,包括零售商店所舉辦的抽獎活動。然而,在PDPC提出新的諮詢指引後,企業可蒐集利用NRIC號碼的情況受到大幅限縮。 由於NRIC號碼與個人資訊息息相關且具不可取代性,無差別地蒐集利用將增加資料被用以從事非法活動之風險,故新的諮詢指引闡明,原則上企業不應蒐集、使用或揭露個人NRIC號碼或複印NRIC,除非有下列兩種例外情況之一:(一)法律要求;(二)為確實證明當事人身分所必要。第一種例外情況,雖因法律要求無須取得當事人同意,但企業仍應踐行告知義務,使當事人知悉NRIC號碼被蒐集、使用或揭露之目的,並確保企業內已採行適當安全措施,防止NRIC號碼被意外洩漏。第二種例外情況則仍須就NRIC號碼的蒐集、使用或揭露取得當事人同意,除非符合個人資料保護法規定下毋庸取得當事人同意之例外(如急救等緊急狀況)。 此外,PDPC針對得蒐集、使用或揭露NRIC號碼或複印NRIC的情況,以情境案例方式於諮詢指引中說明供企業參考,另給予12個月的審視期間,使企業得修正組織內部政策並尋找可行替代方案。
美國NIST發布更新《網路安全資源指南》提升醫療領域的網路安全及隱私風險管理美國國家標準暨技術研究院(National Institute of Standards and Technology, NIST)於2022年7月21日發布更新《網路安全資源指南》(A Cybersecurity Resource Guide, NIST SP 800-66r2 ipd)。本指南源自於1996年美國《健康保險流通與責任法》(Health Insurance Portability and Accountability Act, HIPAA)旨在避免未經患者同意或不知情下揭露患者之敏感健康資料,並側重於保護由健康照護組織所建立、接收、維護或傳輸之受保護電子健康資訊(electronic protected health information, ePHI),包括就診紀錄、疫苗接種紀錄、處方箋、實驗室結果等患者資料之機密性、完整性及可用性。其適用對象包含健康照護提供者(Covered Healthcare Providers)、使用電子方式傳送任何健康資料的醫療計畫(Health Plans)、健康照護資料交換機構(Healthcare Clearinghouses)及為協助上述對象提供健康照護服務之業務夥伴(Business Associate)均應遵守。 本指南最初於2005年發布並經2008年修訂(NIST SP 800-66r1 ipd),而本次更新主要為整合其他網路安全相關指南,使本指南與《網路安全框架》(Cybersecurity Framework, NIST SP 800-53)之控制措施等規範保持一致性。具體更新重點包括:(1)簡要概述HIPAA安全規則;(2)為受監管實體在ePHI風險評估與管理上提供指導;(3)確定受監管實體可能考慮作為資訊安全計畫的一部分所實施的典型活動;(4)列出受監管實體在實施HIPAA安全規則之注意事項及其他可用資源,如操作模板、工具等。特別在本指南第三章風險評估與第四章風險管理提供組織處理之流程及控制措施,包括安全管理流程、指定安全責任、員工安全、資訊近用管理、安全意識與培訓、應變計畫、評估及業務夥伴契約等。而在管理方面包括設施權限控管、工作站使用及安全、設備媒體控制;技術方面則包含近用與審計控管、完整性、個人或實體身分驗證及傳輸安全。上述組織要求得由政策、程序規範、業務夥伴契約、團體健康計畫所組成,以助於改善醫療領域的網路安全及隱私保護風險管理。預計本指南更新將徵求公眾意見至2022年9月21日止。
美國FDA擬修法調整臨床實驗知情同意義務之豁免標準美國FDA擬修法調整臨床實驗知情同意義務之豁免標準 資訊工業策進會科技法律研究所 蔡宜臻法律研究員 2018年11月27日 壹、事件摘要 知情同意(informed consent)是人體試驗受試者保護重要的一環,同時也是生物醫學長期以來的研究傳統,然其規範內容卻會因科技與研究方式的改變而略有調整。美國食品藥物管理局(Food and Drug Administration, FDA)於2018年11月15日發布一份法規提案(proposed rule),公開徵求意見評論。該提案目的在於調整FDA知情同意的相關規定,未來FDA希望允許人體試驗倫理委員會(Institutional Review Boards, IRB)在試驗僅有最小風險(minimal risk)的情況下,得以裁決一臨床實驗案可豁免知情同意的責任,或更改某些「告知要項」[1]。本次法規提案徵詢終止日為2019年1月14日,FDA並規劃於本法規命令正式公告施行後,廢止其於2017年7月所發佈之《IRB豁免或變更臨床實驗之知情同意指南》(IRB Waiver or Alteration of Informed Consent for Clinical Investigations Involving No More Than Minimal Risk to Human Subjects)[2] 貳、重點說明 目前FDA僅允許在危及生命[3]或緊急研究(emergency research)[4]的情況下,得以例外不必符合知情同意的一般要求(general requirements)[5]。而根據FDA於2018年11月15日發布於聯邦公報(Federal Register)的法規提案內容,FDA打算新增「試驗僅有最小風險」(The research involves no more than minimal risk to subjects)做為豁免或變更知情同意項目的甄別標準之一。如此一來若是修法通過,FDA對於知情同意豁免與否的認定標準就會跟1991年制訂的《美國聯邦受試者保護通則》(Federal Policy for the Protection of Human Subjects,簡稱the Common Rule)[6]更加接近。換言之,未來修法通過後,由FDA管理的人體臨床實驗將有三種情形得以豁免或變更知情同意義務:危及生命、緊急研究與僅具有最小風險的研究。 所謂最小風險,係指「研究中預期的傷害或不適的概率和程度,不大於在日常生活中或在進行常規身體或心理檢查時通常遇到的傷害或不適」[7],比如:不需新藥研究申請(investigational new drug application, IND)的新藥研究;醫療器材臨床試驗豁免(investigational device exemption, IDE)之醫療器材研究;檢體之取得為無創(受試者之頭髮或指甲)的臨床研究;為研究目的而蒐集聲音、影片、數據或圖像紀錄;研究個體或群體的特徵或行為;個人或焦點團體訪談等質性研究[8]。FDA指出本次法規提案當中所指的最小風險定義與其附隨條件將與《美國聯邦受試者保護通則》自1991年施行以來之規定一致,即該研究只要同時符合以下四點便可望由IRB審查豁免或變更知情同意義務[9]: 僅有最小風險的研究[10]。 若不豁免或變更知情同意義務,則研究無法順利進行[11]。 不造成受試者權利跟福祉之負面影響[12]。 受試者將在適當時機獲悉進一步研究資訊[13]。 此次提案的法源依據是2016年通過的《21世紀治癒法》(21st Century Cures Act)第3024節所修正之《聯邦食品藥物化妝品法》(Federal Food, Drug, and Cosmetic Act)第505(i)(4)、520(g)(3)節。《21世紀治癒法》第3024節賦予FDA權力放寬臨床實驗的知情同意義務,其立法背景是由於目前FDA相關規範對知情同意要求相對嚴格,當研究者無法滿足現有法規對於知情同意的要求,便可能使潛在的有價值的研究被迫停止[14];又或在某些情形下,要求研究者在進行臨床實驗時取得研究對象的知情同意並不切實際[15]。《21世紀治癒法》通過後,FDA隨即於2017年7月發布《IRB豁免或變更臨床實驗之知情同意指南》,當中指出FDA並不打算在僅有最小風險的臨床研究中,反對IRB做出豁免或變更知情同意項目的判定,若本次法規提案後續正式生效,FDA便會廢止此指南,使其轉為FDA規則(regulation)。 參、事件評析 知情同意是生物醫學研究的學術傳統,包含兩大重點,一是令研究對象充分知悉其所參與的研究,包含其研究目的、內容、風險與預期利益;二是確保研究對象在做出同意或不同意之意思表示時,其意思表示之真實性,由此保障受試者的自主權[16]。 知情同意之概念最早源自1947年的紐倫堡法典(Nuremburg Code),其規範內涵在過去數十年間因為生物醫學的研究方法與進行模式的變革而產生變化。早年的臨床研究主要由政府資助、在單一的機構進行,涉及的受試者人數相對有限;而近三、四十年,醫學研究漸漸發展成多機構、多中心甚至跨國的研究案,受試者可能高達數萬甚至數十萬,同時也逐漸形成跨領域的研究轉型,涉及如社會學、心理學、教育、環境、氣候等學科。在此情形下,研究方法與資料取得勢必與過去截然不同,傳統的知情同意的制度漸漸無法滿足現代醫學研究的需要。1978年貝爾蒙特報告(Belmont Report)便強調應評估臨床研究的風險是否超過日常可接受範圍[17],1981年美國據此制定《美國衛生及公共服務部人體研究保護政策最終規則》(Final regulations amending basic HHS policy for the protection of human research subjects)[18]便首次將「不超過日常風險的臨床實驗」[19]納為知情同意之豁免或變更之標準;1991年制定的《美國聯邦受試者保護通則》亦延續此概念並進一步做出更明確定義(見前述),惟當時FDA基於其業務為確保藥品、生物製劑以及醫療器材安全與執照核發,與《美國聯邦受試者保護通則》作為拘束十六個聯邦機關的一般性規範不同,因此未將「僅有最小風險的臨床實驗」納為豁免或變更知情同意義務的標準[20]。 時序進展至今,資通訊技術的進步所累積的巨量資料逐漸成為生醫研究的重要研究資源,面對這項轉變與研究者對於倫理審查委員會專業性的質疑,美國近年再度嘗試調整修法。2016年通過之《21世紀治癒法》便要求FDA將「僅有最小風險的臨床實驗」納為得豁免或變更免除知情同意的標準之一,可被視為是期望FDA向更為寬鬆的《美國聯邦受試者保護通則》靠攏;另方面,2017年修訂《美國聯邦受試者保護通則》之最終規則(final rule,將於2019年1月生效),也新增「若是研究涉及取得可識別的個人資料或可識別的生物標本,需要證明若無這些資料研究將無法進行」[21],作為豁免或變更知情同意義務的要件,許是為避免個人資料因知情同意的放寬而有遭受濫用之虞。不過這項要件在本次FDA法規提案並未提及。 綜上述,本文整理兩大爭點: 一、最小風險判定標準之不確定性。 最小風險之定義雖明確指「研究中預期的傷害或不適的概率和程度,不大於在日常生活中或在進行常規身體或心理檢查時通常遇到的傷害或不適」[22],惟最小風險之判定仍存在不確定空間。FDA雖強調將承繼《美國聯邦受試者保護通則》自1991年施行以來個案累積之最小風險判定標準,但此一不確定性直接影響的是受試者的自主權,侵害美國憲法所保障的人權精神;此外,也有批評指出FDA所援引的《美國聯邦受試者保護通則》對於最小風險的定義文字過於模糊,容易造成誤解或誤判[23][24][25]。 二、本次法規提案並未新增《美國聯邦受試者保護通則》即將於2019年1月生效的項目,或再度造成FDA規定與其他聯邦機構未能一致的情形。 FDA本次法規提案新增「最小風險」的其中一個原因便是希望盡可能與《美國聯邦受試者保護通則》標準一致。然令人困惑的是,其並未新增《美國聯邦受試者保護通則》即將於2019年1月實施的豁免或變更知情同意義務的要件:「若是研究涉及取得可識別的個人資料或可識別的生物標本,需要證明若無這些資料研究將無法進行」[26]。換言之,即便此次修法提案通過,依舊與會與《美國聯邦受試者保護通則》有落差。更甚者,《美國聯邦受試者保護通則》所新增的要件,實意在保障個人資料不會因知情同意的豁免範圍改變而遭到恣意使用或揭露,有助於保護個人隱私與資料自主,而FDA並未將其納入法規提案內容,或可能造成個資保護之漏洞。此項缺失FDA於法規提案當中亦有提及,或可期待後續修正[27]。 肆、結語 FDA原有關於豁免或變更知情同意的規定與《美國聯邦受試者保護通則》存有寬嚴程度落差,FDA此前僅限定在有生命危險與緊急研究的情形方可為之;而《美國聯邦受試者保護通則》由於是一種一般性規範,所以保障程度較為寬鬆。FDA本次修法將使部分僅有最小風險的臨床實驗可以更為順利進行,同時也使FDA知情同意的規範更加接近當前《美國聯邦受試者保護通則》的規定。惟最小風險的認定存在不確定性,其所可能侵害的是受試者自主權,不可不慎。又,《美國聯邦受試者保護通則》即將在2019年1月規定研究蒐集之個人資料必須對研究有絕對必要方可,而本次FDA的法規提案未見跟進此一新增要件。由於本提案仍在意見評論階段,是以FDA後續是否再度更新提案內容,值得後續關注。 [1] Institutional Review Board Waiver or Alteration of Informed Consent for Minimal Risk Clinical Investigation, 83 Fed. Reg. 57378-57386(Nov. 15, 2018) https://www.federalregister.gov/documents/2018/11/15/2018-24822/institutional-review-board-waiver-or-alteration-of-informed-consent-for-minimal-risk-clinical (last visited Nov. 26, 2018) [2] FOOD AND DRUG ADMINISTRATION[FDA], FDA In Brief: FDA takes steps to allow greater flexibility for clinical investigators about informed consent in minimal risk situations.(2018/11/13) https://www.fda.gov/NewsEvents/Newsroom/FDAInBrief/ucm625747.htm (last visited Nov. 26, 2018) [3] 21 CFR 50.23 [4] 21 CFR 50.24 [5] 有關更多FDA豁免告知同意之項目類別與細部說明,可參考https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=50.23; https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=50.24 (last visited Jan. 8, 2019) [6] 45 CFR 46, subpart A. [7]“the probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests.” (46 CFR 102(i); 21 CFR 50.3(k); 21 CFR 56.102(i)). [8] U.S. DEPARTMENY OF HEALTH & HUMAN SERVICES [HHS], OHRP Expedited Review Categories.(1998) https://www.hhs.gov/ohrp/regulations-and-policy/guidance/categories-of-research-expedited-review-procedure-1998/index.html (last visited Nov. 26, 2018) [9] 45 CFR 46.116 [10] “The research involves no more than minimal risk to subjects” [11] “The research could not be carried out practicably without the waiver or alteration” [12] “The waiver or alteration will not adversely affect the rights and welfare of the subjects” [13] “Where appropriate, the subjects will be provided with additional information about their participation” [14] FOOD AND DRUG ADMINISTRATION[FDA], FDA In Brief: FDA takes steps to allow greater flexibility for clinical investigators about informed consent in minimal risk situations.(2018/11/13) https://www.fda.gov/NewsEvents/Newsroom/FDAInBrief/ucm625747.htm (last visited Nov. 26, 2018) [15] id. [16] 陳子平,〈醫療上「充分說明與同意」之法理在刑法上的效應(上)〉,《月旦法學雜誌》,第278期,頁224(2010)。 [17] THE NATIONAL COMMISSION FOR THE PROTECTION OF HUMAN SUBJECTS OF BIOMEDICAL AND BEHAVIORAL RESEARCH, The Belmont Report—Ethical Principles and Guidance for the Protection of Human Subjects of Research(1978), https://videocast.nih.gov/pdf/ohrp_appendix_belmont_report_vol_2.pdf (last visited Jan. 9, 2019) [18] Final regulations amending basic HHS policy for the protection of human research subjects. 46(16) Fed. Reg. 8366–8391 (Jan. 26, 1981) [19] “those risks encountered in the daily lives of the subjects of the research” (46(16) FR 8373) [20] NATIONAL CENTER FOR BIOTECHNOLOGY INFORMATION[NCBI], Determining Minimal Risk in Social and Behavioral Research(2014), https://www.ncbi.nlm.nih.gov/books/NBK217976/ (last visited Jan. 9, 2019) [21]“if the research involves using identifiable private information or identifiable biospecimens, the research could not practicably be carried out without using such information or biospecimens in an identifiable format” (45 CFR 46.116(f)(3)(iii)) [22] 21 CFR 50.3(k), 56.102(i) [23] Regulations.gov, https://www.regulations.gov/document?D=FDA-2018-N-2727-0010 (last visited Dec. 20, 2018) [24] Shah S, Whittle A, Wilfond B, Gensler G & Wendler D., How do institutional review boards apply the federal risk and benefit standards for pediatric research, JOURNAL OF THE AMERICAN MEDICAL ASSOCIATION, 291(4), 476–482(2004). [25] Lidz C & Garverich S., What the ANPRM missed: Additional needs for IRB reform. JOURNAL OF LAW, MEDICINE AND ETHICS, 41(2), 390–396(2013). [26] 45 CFR 46.116(f)(3)(iii) [27] Supra note No. 1