世界經濟論壇發布《人工智慧公平性和包容性藍圖》白皮書

世界經濟論壇(World Economic Forum, WEF)於2022年6月29日發布《人工智慧公平性和包容性藍圖》白皮書(A Blueprint for Equity and Inclusion in Artificial Intelligence),說明在AI開發生命週期和治理生態系統中,應該如何改善公平性和強化包容性。根據全球未來人類AI理事會(Global Future Council on Artificial Intelligence for Humanity)指出,目前AI生命週期應分為兩個部分,一是管理AI使用,二是設計、開發、部署AI以滿足利益相關者需求。
包容性AI不僅是考量技術發展中之公平性與包容性,而是需整體考量並建立包容的AI生態系統,包括(1)包容性AI基礎設施(例如運算能力、資料儲存、網路),鼓勵更多技術或非技術的人員有能力參與到AI相關工作中;(2)建立AI素養、教育及意識,例如從小開始開啟AI相關課程,讓孩子從小即可以從父母的工作、家庭、學校,甚至玩具中學習AI系統對資料和隱私的影響並進行思考,盡可能讓使其互動的人都了解AI之基礎知識,並能夠認識其可能帶來的風險與機會;(3)公平的工作環境,未來各行各業需要越來越多多元化人才,企業需拓寬與AI相關之職位,例如讓非傳統背景人員接受交叉培訓、公私協力建立夥伴關係、提高員工職場歸屬感。
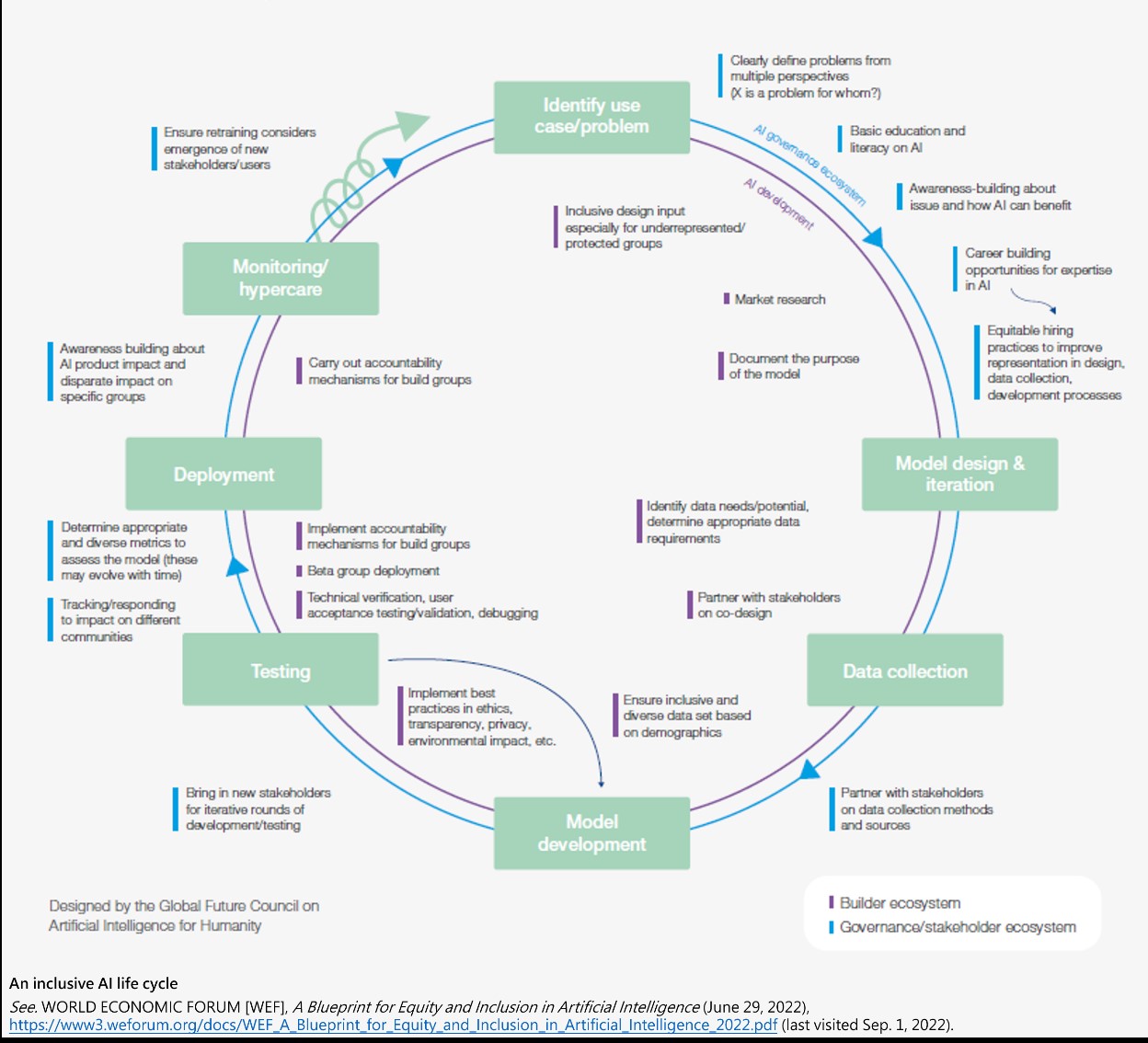
在設計包容性方面,必須考慮不同利益相關者之需求,並從設計者、開發者、監督機關等不同角度觀察。本報告將包容性AI開發及治理整個生命週期分為6個不同階段,期望在生命週期中的每個階段皆考量公平性與包容性:
1.了解問題並確定AI解決方案:釐清為何需要部署AI,並設定希望改善的目標變量(target variable),並透過制定包容性社會參與框架或行為準則,盡可能實現包容性社會參與(特別是代表性不足或受保護的族群)。
2.包容性模型設計:設計時需考慮社會和受影響的利益相關者,並多方考量各種設計決策及運用在不同情況時之公平性、健全性、全面性、可解釋性、準確性及透明度等。
3.包容性資料蒐集:透過設計健全的治理及隱私,確定更具包容性的資料蒐集路徑,以確保所建立之模型能適用到整體社會。
4.公平和包容的模型開發及測試:除多元化開發團隊及資料代表性,組織也應引進不同利益相關者進行迭代開發與測試,並招募測試組進行測試與部署,以確保測試人群能夠代表整體人類。且模型可能隨著時間發展而有變化,需以多元化指標評估與調整。
5.公平地部署受信任的AI系統,並監控社會影響:部署AI系統後仍應持續監控,並持續評估可能出現新的利益相關者或使用者,以降低因環境變化而可能產生的危害。
6.不斷循環發展的生命週期:不應以傳統重複循環過程看待AI生命週期,而是以流動、展開及演變的態度,隨時評估及調整,以因應新的挑戰及需求,透過定期紀錄及審查,隨時重塑包容性AI生態系統。
綜上,本報告以包容性AI生態系統及生命週期概念,期望透過基礎設施、教育與培訓、公平的工作環境等,以因應未來無所不在的AI社會與生活,建立公司、政府、教育機構可以遵循的方向。
- World Economic Forum [WEF], A Blueprint for Equity and Inclusion in Artificial Intelligence (June 29, 2022)
- A Blueprint for Equity and Inclusion in Artificial Intelligence, World Economic Forum (June 29, 2022)
- Global Future Council on Artificial Intelligence for Humanity, World Economic Forum (June 29, 2022)

許嘉芳
法律研究員 編譯整理
World Economic Forum [WEF], A Blueprint for Equity and Inclusion in Artificial Intelligence (June 29, 2022), https://www3.weforum.org/docs/WEF_A_Blueprint_for_Equity_and_Inclusion_in_Artificial_Intelligence_2022.pdf (last visited Sep. 1, 2022).
A Blueprint for Equity and Inclusion in Artificial Intelligence, World Economic Forum (June 29, 2022), https://www.weforum.org/whitepapers/a-blueprint-for-equity-and-inclusion-in-artificial-intelligence (last visited Sep. 1, 2022).
Global Future Council on Artificial Intelligence for Humanity, World Economic Forum (June 29, 2022), https://www.weforum.org/communities/gfc-on-artificial-intelligence-for-humanity (last visited Sep. 1, 2022).
王柏霳,〈美國國家標準暨技術研究院規劃建立「人工智慧風險管理框架」,並徵詢公眾對於該框架之意見〉,2021年10月, https://stli.iii.org.tw/article-detail.aspx?tp=1&d=8727&no=64(最後瀏覽日:2022/09/02)。

 科法觀點
科法觀點

為了持續維持日本國內以及與東京奧運舉辦相關的關鍵基礎設施服務的安全性,日本內閣網路中心於2017年4月19日公布關鍵基礎設施資訊安全對策第4次行動計畫。 在第4次行動計畫,關鍵基礎設施防護目的主要是以關鍵基礎設施的功能保證為考量,盡量減少關鍵基礎設施IT故障的發生,並提升從事故中恢復的速度。因此,第4次行動計畫除持續檢討並改善第3次行動計畫原有政策外,較重要的變革為OT(Operation Technology)的重視與風險對應機制整備。在安全基準整備與落實情況方面,要求關鍵基礎設施產業須將OT的觀點融入人才培育。在資訊分享制度方面,分享的資訊範圍應包含IT、OT與IoT的資訊,並排除資訊分享的障礙。而在風險管理部分,日本從功能保證的觀點出發,新增風險情況對應準備的要求,包含事業持續計畫的提出與緊急應變措施的制定等。而在防護基礎強化上,該行動計畫認為關鍵基礎設施產業的IT、OT人員及法務部門必須依其內部資訊安全策略共同為關鍵基礎設施安全而跨組織合作。 另外,第4次行動計畫變更電力領域關鍵基礎設施的重要系統,從原有的運轉監視系統變更為智慧電表,以及新增化學、信用卡與石油三大關鍵基礎設施領域的業者、關鍵系統與因IT故障對關鍵基礎設施可能造成的危害影響。
美國政府於2014年初提出幾點重要聲明,加強改善國家專利品質美國總統歐巴馬於2014年初對於美國專利改革及產業創新的規範做進一步的聲明。美國近年來針對專利法改革有許多大規模的法案實施,目的希望能提升整體美國產業,包括2011年通過的美國發明法案(Leahy-Smith America invents Act, AIA),目的希望能讓美國專利系統更加完善,保護專利權人及促進產業創新等目的。然許多專利仍被NPE或是專利蟑螂控訴侵權,反而讓專利權被用來當做專利訴訟的一個工具,花費更多的經費在訴訟及和解上,有違當初白宮要進行專利改革的初衷。 因此歐巴馬在年初為了能鼓勵創新及增加專利系統的品質而發布幾點執行聲明(executive actions): 1、著重prior art的檢索:USPTO開始著重prior art的搜尋,幫助專利審查能更詳盡。 2、增進專利審查人的技術訓練:提供教育專業訓練,讓專利審查人能隨時更新最新的技術,能在審查過程中對於技術上的認知能更專業。 3、Pro brono幫助:USPTO提供pro brono的幫助。許多發明人對於如何申請專利及如何使其專利被妥善保護等規範較缺乏相關資訊、或沒有資金聘請顧問協助此方面保護,因此USPTO會提供教育及實務訓練,讓這些較小的公司或資源較缺乏之發明人的專利得以獲得保護。
英國科學辦公室發布分佈式分類帳技術報告,提出八大建議2016年1月, 隸屬英國商業、創新和技術部 (Department for Business, Innovation and Skills,BIS)的科學辦公室(Government Office for Science)發布「分佈式分類帳技術:區塊鏈以外(Distributed Ledger Technology:beyond block chain)」研究報告。本篇報告由產官學界合作完成,主要在評估分佈式分類帳技術可以運用在哪一些公私領域,並決定政府以及私人應該採取哪些行動以促進分佈式分類帳技術可被有益運用,並避免可能帶來的傷害。 該份研究報告認為,分佈式分類帳技術可在多個領域協助政府機構,包含徵稅、提供福利、發行護照、土地登記、確保商品供應鏈並且確保政府記錄與服務的完整性。相較於其他網路系統,分佈式分類帳技術較不易受駭客攻擊,而且由於每個参與者都有一份帳簿副本,如果有惡意竄改的狀況,也可以輕易被發現,但這不表示分佈式分類帳技術就不會被駭客攻擊。 數位五國(Digital 5,D5)之一的愛沙尼亞,已多年實驗運用分佈式分類帳技術於公領域服務多年。愛沙尼亞政府透過私人公司運用分佈式分類帳技術建制「免金鑰簽名設施(Keyless Signature Infrastructure,KSI)」,KSI允許愛沙尼亞公民驗證其在政府資料庫資訊的完整性,並避免內部人透過政府網路從事非法活動。KSI確保公民資訊安全以及準確,因而可協助愛沙尼亞政府提供數位化的公司登記以及稅務服務,減少政府以及社會大眾的行政作業負擔。 除此之外,分佈式分類帳技術也有助於確保商品以及智慧財產權的所有以及出處。例如Everledger此一系統可用於確保鑽石的身分,從礦產、切割到銷售,可減少並避免欺詐以及「血鑽石」進入市場。 簡而言之,分佈式分類帳技術提供政府可減少詐欺、腐敗、錯誤以及紙上作業成本的框架,並透過資訊分享、公開透明以及信任,具有可重新定義政府與公民關係的潛力。對於私領域而言也具有同樣可能性,報告特別提出可透過分佈式分類帳技術發展「智慧契約」,可增加信任度並提高效率。據此,本報告針對政府部門提出八大建議: (1) 應成立專責部門,並與產業、學界緊密合作,並應考慮成立臨時性的專家諮詢團隊。 (2) 英國的研究社群應該要投入研究確保分佈式分類帳技術具備可即性、安全性以及內容準確性。 (3) 政府應支持為地方政府成立分佈式分類帳技術實地教學者,匯聚所有測試技術以及其運用的所需元素。 (4) 政府需要思考如何為分佈式分類帳技術建立妥適的法制框架。法規需要配合新科技應用技術的發展而進步。 (5) 政府應該與產學合作確保相關標準可以符合分佈式分類帳技術及其內容完整性、安全性以及隱私的需求。 (6) 政府應與產學合作確保最有效率以及最可用的身分認證網路協議可為個人及組織所使用,這項工作應與國際標準的發展與執行緊密連結。 (7) 政府應對分佈式分類帳技術進行試驗,以評估該項技術在公領域的可行性。 (8) 建議成立跨部門的利益群體,結合分析以及政策群體,以生成並發展潛在使用案例,並且在公民服務中提供具備知識的專家人員。 除了八大建議,管理與法制上,本報告指出分佈式分類帳技術具有兩種管理規範:法律規範以及技術規範。法律規範是「外部」規範,法律規範可能會被違反,緊接著面臨違法處罰的問題。技術規範是「內部」規範,假如違反技術規範,「錯誤(error)」產生無法運作,因此「規範」本身就可以確保會被遵循。換句話說,技術規範可以節省法律規範的執法成本。另外一方面,分佈式分類帳技術為去中心化技術,如果要以法制管理,也只能在参與者身上施加法律義務,例如Bitcoin,只能對於提供Bitcoin交易服務的平台施加法律義務。美國紐約州金融服務部所發行的比特幣交易執照BitLicnese即為一例。因此,基於去中心化的特性,報告建議政府單位應該要儘量参與技術標準的制定,並且配合技術標準制定相關法律,法律規範與技術規範兩者應該要交互影響。
從交易成本概念談智慧財產資訊揭露的原則與效益